一种水稳定微孔双功能MOFs材料及其制备方法和用途
一种水稳定微孔双功能mofs材料及其制备方法和用途
技术领域
1.本发明属于多功能mofs材料制备技术领域,具体涉及一种以3-羟基-1,2,4,5-苯四羧酸配体(h6obtec)为底物的微孔水稳定mof材料,[zn6(obtec)2(h2o)6]
·
8h2o,及其制备方法和作为染料吸附剂或金属离子探针的用途。
背景技术:
[0002]
随着人类纺织、皮革、造纸、印染、塑料、冶炼、电镀等工业的快速发展,使得水资源的污染也日益加剧,受污水质的污染物成分也更加复杂,其中由有机染料和重金属离子所带来的污染最为严重。大多数的有机染料分子和重金属离子的毒性很大,致癌性很高。金属离子特别是重金属离子的超标对水体中生物的生存造成了严重威胁。当重金属离子进入生物的体内时,会在生物体内累积,引起动物的畸形,并且可能通过食物链的作用进入到人体内,威胁人类的生命健康。由于染料分子对水质的颜色影响很大也倍受人们广泛的关注。为了人体健康和人类社会的可持续发展,检测和除去废水中的染料分子和重金属离子尤为重要。
[0003]
合成染料一般具有氮杂的芳香结构,因此往往具有较高的稳定性,难从废水中除去它们,目前,染料废水常见的处理方法主要包括絮凝法、吸附法、电化学法、氧化法、生物法以及生物膜法等。相对而言,由于染料可生化能力较差,生物处理法往往效率较低。而生物膜渗透法、反渗透法、化学沉淀法、超滤渗透、光解等等方法往往处理范围较小,成本较高。而吸附法中的物理吸附法具有操作过程简单、效率高等优点,是处理染料废水的最有效方法之一。传统的吸附剂材料如活性炭或者生物炭等等往往孔洞较小而且规则性较差,有效活性位点的数量较低,它们在染料吸附方面往往达不到预期的效果。因此,开发吸附性能优异的染料吸附剂材料仍然是一项持续的工作。
[0004]
mofs材料是有机配体和金属离子或金属离子形成的多核团簇构筑的多孔化合物。通过设计不同的配体,可以有效调控吸附材料孔径,达到选择性吸附的目的,另外一方面,如果所产生mofs含有不饱和金属配位点,或者是配体含有未配位活性基团,将在一定程度上增强吸附性能,甚至达到选择性吸附分离的效果。当mofs作为吸附材料使用时,mofs材料可能与有机染料分子之间产生静电相互作用、π-π堆积作用、氢键作用、疏水作用、酸碱相互作用等等,这些都会显著影响mofs材料对吸附质的吸附,甚至可能决定材料的吸附性能。但吸附有机染料时,由于不同染料分子结构、尺寸、官能团的不同,可能导致不同mofs材料对不同染料的吸附性能产生差异,这为设计选择吸附某种特定的染料的mofs材料提供了可能。
[0005]
在另外一个方面,mofs材料也被认为是最有前途的化学传感检测材料之一。mofs材料作为一种比表面积大吸附能力强的物质,与分子或离子作用时,往往能够吸附相应的分子和离子,使其在mofs材料的孔洞内或表面聚集,产生各种相互作用,从而由此增强mofs材料的传感信号强度,使得mofs作为传感器材料在检测被分析物时具有更低的检测极限和更高的灵敏度。和吸附类似,有目的制备含有不饱和金属配位点以及未配位基团,增强mofs
与被检测物质相互作用,从而达到更好的检测效果。
[0006]
mofs材料的制备方法有很多,主要包括水热、溶剂热、离子热、微波合成法等等。尽管mofs材料合成方法多种多样,但是溶剂热法使用较多。而溶剂热法会涉及使用一些高沸点的机溶剂,如dmf、dma、dme等等,这些溶剂本身难以降解,合成过程不仅会对环境造成二次污染。而且这些材料在水中往往具有较差的稳定性,这就限制了其在水质环境中的使用,如文献广泛引用的mof-5由于在水中的稳定性较差,客观上就限制了其在污水处理中的应用。
技术实现要素:
[0007]
本发明提供了一种染料吸附剂的制备方法。本发明的目的在于提供一种工艺简便,制备过程污染小、通过以3-羟基-1,2,4,5-苯四羧酸配体为底物构筑水稳定mofs,可以作为染料吸附剂和al
3+
检测荧光探针。
[0008]
本发明有目的的利用3-羟基-1,2,4,5-苯四羧酸,和zn
2+
作用,通过控制反应条件,在水热法条件下,合成了一种以微孔水稳定配合物[zn6(obtec)2(h2o)6]
·
8h2o,该配合物具有的一维孔道。而且,该配合物的不饱和金属配位点很容易和阴离子作用,从而增强其对阳离子染料亚甲基蓝的吸附性能,由于尺寸大小对中性染料结晶紫吸附能力较弱。与此同时,悬浊溶液中加入不同的金属离子,溶液的荧光发射强度却有明显的改变,当配合物悬浊液中加入al
3+
后溶液的荧光发射强度明显增强(约4倍);但当加入co
2+
、cu
2+
、fe
3+
、ni
2+
离子时,配合物的荧光发光强度明显降低,发生了荧光淬灭,并且淬灭率都在95%以上,可以作为al
3+
检测荧光探针。同时,该配合物是利用水做溶剂制备的一种水稳定的微孔材料,制备过程不使用有机溶剂,不会对环境造成二次污染,是一种具有应用前景的新型染料吸附剂材料。
[0009]
本发明的一种基于3-羟基-1,2,4,5-苯四羧酸构筑的水稳定染料吸附剂,zn离子和有机配体3-羟基-1,2,4,5-苯四羧酸(h6obtec)在碱性条件下,控制水热反应制备而得。其化学式可表示为[zn6(obtec)2(h2o)6]
·
8h2o;
[0010]
从连接构筑的角度,每个晶体学不对称结构单元中包含了三个晶体学独立的zn(ⅱ)原子,一个完全去质子化的obtec
6-配体,三个配位水分子和四个游离水分子,其中zn(3)和zn(4)占有率为0.5,四种zn原子,其中zn(1),zn(2)以及zn(3)均为六配位,形成变形的八面体构型;zn(4)为两配位环境,除了和羟基氧原子作用,还与另外一个配体苯环脱氢碳原子作用,并呈现出v形的几何构型;zn-o键长范围在o键长范围在zn(1),zn(2)以及zn(3)通过羧基桥联氧原子连接成一维zn-o带状结构,所述一维zn-o带状结构与obtec
6-配体桥联形成一个沿a轴方向的的一维孔洞。
[0011]
从骨架连接构筑角度,配合物[zn6(obtec)2(h2o)6]
·
8h2o属于单斜晶系,空间群为p21/c。晶胞参数为α=90
°
,β=113.1510(10)
°
,γ=90
°
[0012]
本发明水稳定微孔双功能mofs材料的合成方法,其步骤如下:
[0013]
步骤1:准确称量有机配体h6obtec和二乙胺溶液,加入去离子水,于样品瓶中超声混合,超声混合均匀后得混合溶液1备用;所述h6obtec、二乙胺与去离子水的摩尔比为1:
3.5:5500。
[0014]
步骤2:准确移取zn(no3)2·
3h2o溶液逐滴加入步骤1样品瓶中的混合溶液1中,超声溶解混合均匀后静止待用,为混合溶液2;其中zn(no3)2·
3h2o与有机配体h6obtec的摩尔比例为3:1。
[0015]
步骤3:首先将步骤2中的装有混合溶液2的样品瓶放入具有聚四氟乙烯内衬的不锈钢釜中,然后将反应釜放入100-120℃烘箱中,保持4天,降温1天,取出后用去离子水洗涤,过滤后去离子水洗涤自然风干,得到无色晶体。
[0016]
利用单晶衍射仪(ccd)、x射线衍射(xrd)、热重(tg)等对产物的结构与性质进行分析。
[0017]
所述前驱配体3-羟基-1,2,4,5-苯四羧酸,以2,3,5,6-四甲基溴苯为原料,通过氧化、碱熔、酸化、重结晶样品抽滤后于60度恒温干燥箱中干燥12小时制备的。
[0018]
将本发明制备的一种水稳定微孔双功能mofs材料用于选择性吸附有机染料亚甲基蓝或甲基橙的用途。
[0019]
将本发明制备的一种水稳定微孔双功能mofs材料作为金属al
3+
离子检测荧光探针的用途。
[0020]
本发明的优点:
[0021]
1.本发明采用水热法制备的金属-有机骨架材料,含有因为一维zn-o带,具有良好的水稳定性,合成的过程不使用有机溶剂,不会造成二次污染。
[0022]
2.本发明制备的金属-有机骨架材料中有丰富的配位水分子,可以作为潜在的不饱和活性位点,暴露的不饱和活性点能够与离子型染料分子相互作用而增强染料的吸附性能,配合物良好的荧光性能和特殊zn配位环境为配合物作为分子探针提供了良好的物理环境。
附图说明
[0023]
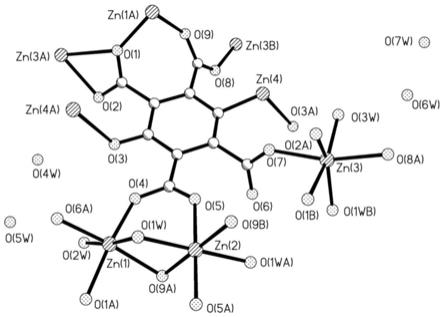
图1为本发明所制备样品配位环境图。
[0024]
图2为本发明所制备样品zn(1)、zn(2)和zn(3)通过羧基氧原子桥联形成一维zn-o带状结构。
[0025]
图3为本发明所制备样品三维框架结构图,从b轴方向可以观察到的一维孔洞结构。
[0026]
图4为本发明所制备样品吸附亚甲基蓝(mb)的紫外吸收曲线。
[0027]
图5为本发明所制备样品吸附结晶紫(cv)的紫外吸收曲线。图6为本发明所制备样品吸附甲基橙(mo)的紫外吸收曲线。
[0028]
图7为本发明所制备样品吸附罗丹明b(rhb)的紫外吸收曲线。
[0029]
图8为本发明样品对四种染料吸附量与时间的关系图。
[0030]
图9为金属离子对本发明样品配合物荧光发光强度的影响柱状图。
具体实施方式
[0031]
下面结合说明书附图和具体实施例对本发明作进一步说明,但本发明不限于以下实例。
[0032]
实施例1
[0033]
准确称取h6obtec 13.5mg(0.05mmol),然后加入到10ml的玻璃瓶(样品瓶)中;然后加入5ml去离子水和1mol/l的二乙胺溶液175ul(0.175mmol),超声至有机配体完全溶解后,再加入1mol/l的zn(no3)2·
6h2o 0.15mmol,再次超声至混合均匀。最后将样品瓶放入含有25ml聚四氟乙烯内衬的钢釜中,将钢釜在120℃烘箱中保持96小时,降温24小时冷却至室温后,将产物用去离子水洗涤3次,风干,得到无色透明晶体,产率约65%(以h6obtec计算)。
[0034]
实施例2:反应温度由实施例1的120℃变为110℃。
[0035]
准确称取h6obtec 13.5mg(0.05mmol),然后加入到10ml的玻璃瓶(样品瓶)中;然后加入5ml去离子水和1mol/l的二乙胺溶液175ul(0.175mmol),超声至有机配体完全溶解后,再加入1mol/l的zn(no3)2·
6h2o 0.15mmol,再次超声至混合均匀。最后将样品瓶放入含有25ml聚四氟乙烯内衬的钢釜中,将钢釜在110℃烘箱中保持96小时,降温24小时冷却至室温后,将产物用去离子水洗涤3次,风干,得到无色透明晶体,但相比实例1,实例2产率约60%。
[0036]
实施例3:反应温度由实施例1的120℃变为100℃。
[0037]
准确称取h6obtec 13.5mg(0.05mmol),然后加入到10ml的玻璃瓶(样品瓶)中;然后加入5ml去离子水和1mol/l的二乙胺溶液175ul(0.175mmol),超声至有机配体完全溶解后,再加入1mol/l的zn(no3)2·
6h2o 0.15mmol,再次超声至混合均匀。最后将样品瓶放入含有25ml聚四氟乙烯内衬的钢釜中,将钢釜在100℃烘箱中保持96小时,降温24小时冷却至室温后,将产物用去离子水洗涤3次,风干,得到少量无色透明晶体,相比实例2,实例3中有较少的白色絮状杂质。
[0038]
图1为本发明所制备样品配位环境图。其存在四种晶体学独立的zn(ⅱ),其中zn(1)、zn(2)和zn(3)均为六配位形成扭曲的八面体配位环境,而zn(4)为二配位,呈现出v形的几何构型。
[0039]
图2为本发明所制备样品zn(1)、zn(2)和zn(3)通过羧基氧原子桥联形成一维zn-o带状结构。
[0040]
图3为本发明所制备样品三维框架结构图,从b轴方向可以观察到的一维孔洞结构。
[0041]
对比例1:反应温度由实施例1的120℃变为130℃。
[0042]
准确称取h6obtec 13.5mg(0.05mmol),然后加入到10ml的玻璃瓶(样品瓶)中;然后加入5ml去离子水和1mol/l的二乙胺溶液175ul(0.175mmol),超声至有机配体完全溶解后,再加入1mol/l的zn(no3)2·
6h2o 0.15mmol,再次超声至混合均匀。最后将样品瓶放入含有25ml聚四氟乙烯内衬的钢釜中,将钢釜在130℃烘箱中保持96小时,降温24小时冷却至室温后,将产物用去离子水洗涤3次,风干,得到少量无色透明晶体,并有部分黑色白色沉淀。
[0043]
对比例2:与实施例1相比,二乙胺与h6obtec的摩尔比大于4:1
[0044]
准确称取h6obtec 13.5mg(0.05mmol),然后加入到10ml的玻璃瓶(样品瓶)中;然后加入5ml去离子水和1mol/l的二乙胺溶液0.20、0.25mmol,超声至有机配体完全溶解后,再加入1mol/l的zn(no3)2·
6h2o 0.15mmol,再次超声至混合均匀。最后将样品瓶放入含有25ml聚四氟乙烯内衬的钢釜中,将钢釜在120℃烘箱中保持96小时,降温24小时冷却至室温后,将产物用去离子水洗涤3次,风干,得到无色透明晶体,相比实例1,实例5中只有较少量
成型晶体,并有大量不明杂质。
[0045]
对比例3:与实施例1相比,二乙胺与h6obtec的摩尔比小于3:1
[0046]
准确称取h6obtec 13.5mg(0.05mmol),然后加入到10ml的玻璃瓶(样品瓶)中;然后加入5ml去离子水和1mol/l的二乙胺溶液0.15、0.10mmol,超声至有机配体完全溶解后,再加入1mol/l的zn(no3)2·
6h2o 0.15mmol,再次超声至混合均匀。最后将样品瓶放入含有25ml聚四氟乙烯内衬的钢釜中,将钢釜在120℃烘箱中保持96小时,降温24小时冷却至室温后,将产物用去离子水洗涤3次,风干,得到无色透明晶体,相比实例1,实例6中只有较极少量成型晶体,并产生部分不明物质。
[0047]
对比例4:由实施例1的二乙胺变为naoh
[0048]
准确称取h6obtec 13.5mg(0.05mmol),然后加入到10ml的玻璃瓶(样品瓶)中;然后加入5ml去离子水和氢氧化钠(0.175mmol),超声至有机配体完全溶解后,再加入1mol/l的zn(no3)2·
6h2o 0.15mmol,再次超声至混合均匀。最后将样品瓶放入含有25ml聚四氟乙烯内衬的钢釜中,将钢釜在120℃烘箱中保持96小时,降温24小时冷却至室温后,相比实例1,实例7中无目标产物生成,并产生大量黑色和白色不明物质。
[0049]
吸附性能测试
[0050]
测试前首先利用溶剂置换法除去本发明样品孔道结构中存在的客体分子。具体步骤如下,将配合物浸泡于甲醇溶剂中三天,平均每6个小时置换一次甲醇溶剂,最后放置于60℃真空干燥箱中干燥12个小时,活化吸附剂材料。使用实施例1所得样品进行测试,所使用样品吸附剂的质量均为30mg分别对应100ml亚甲基蓝(mb)、结晶紫(cv)、甲基橙(mo)和罗丹明b(rhb)染料溶液(质量浓度均为50mg/l),在常温条件下,测定0h-8h内不同时刻各染料水溶液的吸光度,随着时间的增加,mb(最大吸收波长约为664nm)溶液的吸光度和cv(最大吸收波长约为590nm)溶液的吸光度都下降十分明显,而mo(最大吸收波长约为463nm)溶液的和rhb(最大吸收波长约为552nm)溶液的吸光度则下降较少。
[0051]
图4,图5,图6,图7分别为本发明所制备样品吸附四种有害染料亚甲基蓝(mb),结晶紫(cv),甲基橙(mo)以及罗丹明b(rhb)染料的紫外吸收曲线。该测试说明了,常温条件下,本发明材料吸附剂对mb和mo具有良好的吸附性能,而对rhb和cv的吸附性能较差,表现出一定的选择性。
[0052]
图8为本发明样品对四种染料吸附量与时间的关系图。常温条件下,8h内,本发明材料对亚甲基蓝、甲基橙、罗丹明b和结晶紫的吸附量分别达到了458.7mg/g、296.7mg/g、237.0mg/g和95.3mg/g。进一步说明了本发明材料吸附剂对亚甲基蓝和甲基橙具有较好的吸附能力,对罗丹明b和结晶紫吸附性能较差,表现出一定的选择性。
[0053]
使用实施例1所得样品进行金属离子荧光感应性能测试
[0054]
配合物在进行使用前,需要进行研磨,然后准确称取10mg充分研磨的配合物加入到30ml去离子水中,超声混合均匀。即得到各种配合物的悬浊液。然后将悬浊液进行离心后取出清液待用。配合物的荧光发光测定及荧光感应检测使用的是配合物的上清液。
[0055]
配合物进行荧光检测前,需要使用上清液进行荧光扫描检测各种配合物的最大发射波长。配合物的荧光感应检测方法为:在同体积的上清液中加入相同浓度和相同体积的金属离子溶液,控制金属离子的浓度约为1mm,加入完成后在室温下搅拌10min后,然后在相同激发波长条件下进行荧光发光性能测试。某种金属离子对配合物荧光发光性能的影响关
系测试方法与这种方法类似,不同的是金属离子的浓度存在梯度差异。配合物对金属离子感应的灵敏度通过感应时间的长短来衡量的,方法与上述方法类似,不同的是当加入检测离子后在不同的时间阶段进行荧光发光性能检测,直到荧光强度不发生改变时停止。
[0056]
图9为金属离子对本发明样品配合物荧光发光强度的影响柱状图,结果显示如图9,当配合物悬浊液中加入al
3+
后溶液的荧光发射强度明显增强(约4倍);而co
2+
、cu
2+
、fe
3+
、ni
2+
离子时,配合物的荧光发光强度明显降低,发生了荧光淬灭,并且淬灭率都在95%以上,部分离子变化不大,可以作为al
3+
检测荧光探针。
[0057]
实施例1、实施例2和实施例4均能得到目标产物,其中实施例1中二乙胺和3-羟基-1,2,4,5-苯四羧酸的摩尔比为3.5:1时效果最好,用此比例得到的样品进行单晶测试,表1为其晶体数据和精修参数。
[0058]
表1本发明所制备样品的晶体数据和精修参数
[0059]
[0060]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1