一种制备古古甾酮的方法与流程
1.本发明属于有机化学合成/药物合成技术领域,具体涉及一种制备古古甾酮的方法。
背景技术:
2.古古甾酮也称为没药甾酮、木苦甾酮、香胶甾酮、固甾酮或孕二烯二酮。古古甾酮具有如下化学式1和2所示的两种同分异构体。化学式1为e-4,17(20)-孕二烯-3,16-二酮,也称为e-古古甾酮。化学式2为z-4,17(20)-孕二烯-3,16-二酮,也称为z-古古甾酮。
[0003][0004]
许多研究表明,古古甾酮的生物活性至少部分是由于它们作为farnesoid x受体(fxr)配体的作用。同分异构体1和2都被发现都具有选择性调节fxr基因的表达,特别是正向调节细胞色素的表达cyp7a1,从而诱导胆汁酸中胆固醇的分解代谢,降低胆固醇水平。以前古古甾酮主要从树脂中提取,收率仅1%左右,为了更好地探索古古甾酮的生物活性和药物作用,合成古古甾酮的工作变得非常有意义。
[0005]
gioiello et al./steroids 77(2012)250
–
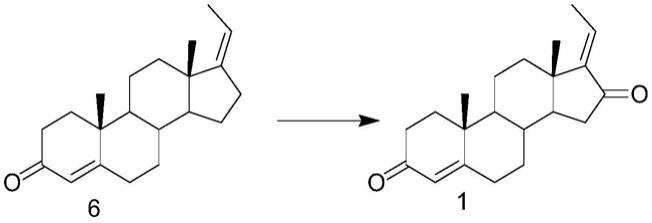
254报道了一种高效的合成方法,见如下化学流程,该方法以式3所示的4-ad(4-雄烯-3,17-二酮)为起始原料,经过3-醚化保护得式4所示化合物,再通过witting反应构建17-侧链得式5所示化合物,脱保护得式6所示化合物,再在16-位引入羟基得式7所示化合物,然后经氧化得到化合物1。该路线简洁而且收率较高,也避免了传统工艺的高温黄鸣龙反应,但是合成下述化合物7引入羟基时通常只有少部分e-古古甾酮生成,需要进一步氧化得到e-古古甾酮。因此使得e-古古甾酮的收率大打折扣。
[0006][0007]
因此,本领域需要一种步骤更为简洁且收率更高的制备古古甾酮的方法。
技术实现要素:
[0008]
因此,本发明提供一种制备古古甾酮的方法,所述古古甾酮为式1所示的4,17-孕二烯-3,16-二酮,所述古古甾酮通过式6所示化合物经一步法制备得到,具体包括将式6所示化合物溶解在有机溶剂中,加入自由基引发剂过氧化苯甲酰和催化剂,通入空气进行氧化反应得到所述古古甾酮,所述有机溶剂中包含c3-c6的酮,所述催化剂包括n-羟基邻苯二甲酰亚胺和过渡金属钴、锰或铜的氧化物或盐。
[0009][0010]
在一种具体的实施方式中,所述过渡金属钴、锰或铜的氧化物或盐为选自二氧化锰、醋酸锰、醋酸钴和碘化亚铜中的一种或多种,优选使用二氧化锰。
[0011]
在一种具体的实施方式中,所述有机溶剂中还包括酯类协同溶剂,优选所述酯类协同溶剂为乙酸乙酯、乙酸丁酯、丙酸乙酯和醋酸异丙酯中的一种或多种。
[0012]
在一种具体的实施方式中,所述一步法制备过程中,反应温度为30~80℃,优选45~65℃。
[0013]
在一种具体的实施方式中,引发剂n-羟基邻苯二甲酰亚胺与式6所示底物的质量比为0.075~0.3:1,优选0.1~0.2:1,过渡金属钴、锰或铜的氧化物或盐与式6所示底物的质量比为0.001~0.1:1,优选0.0015~0.005:1。
[0014]
也就是说,在现有技术中,甾体17-烯的16位氧化时,会引入部分16-酮及部分16-羟基化合物,而合成古古甾酮需要全部引入16-酮,则17-烯的16位需要经过两步氧化(化合物6生成化合物7以及化合物7生成化合物1)才能得到。而本发明利用空气氧化,以过氧化苯甲酰为引发剂,再加入特殊金属氧化物或盐辅助,可一步完成该反应,且收率较高。
[0015]
总的来说,本发明以更有效的方式,直接通过空气氧化引入16-位碳基得到目标化
合物古古甾酮,其步骤更短,收率更高,更环保。
附图说明
[0016]
图1为本发明实施例1中制备得到的古古甾酮的氢谱图。
[0017]
图2为本发明实施例1中制备得到的古古甾酮的质谱图。
具体实施方式
[0018]
在一种具体的实施方式中,所述古古甾酮的制备方法包括如下四个步骤,第1步,由式3所示化合物生成式4所示化合物(3位醚化保护步骤);第2步,由式4所示化合物生成式5所示化合物(17位侧链构建步骤);第3步,由式5所示化合物生成式6所示化合物(3位脱保护步骤);以及最重要的第4步,由式6所示化合物生成式1所示化合物(一步法引入16位羰基的步骤)。
[0019][0020]
最重要的,第4步(化合物6
→
1),其中加入微量过渡金属钴、锰或铜的氧化物或盐,例如mno2,便能取得很好的反应效果。推测其原因,一方面是过渡金属钴、锰或铜与有机化合物配对时能产生特殊的空间效应和电子效应,从而有利于提高氧化反应的选择性,另一方面是过渡钴、锰或铜的氧化物或者盐本身具有一定氧化性。
[0021]
具体地:
[0022]
第1步,化合物3与甲醇或乙醇在脱水剂和催化剂存在下发生醚化反应。脱水剂可以是原甲酸三乙酯、原乙酸三甲酯、原甲酸三甲酯等。反应温度为0~40℃。
[0023]
第2步,化合物4与乙基三苯基卤化膦(ph3p+ch2ch3x-)在无水溶剂中和强碱作用下合成化合物5。ph3p+ch2ch3x-中x可以是氯、溴、碘中的任意一种。溶剂可以是四氢呋喃、甲醇、乙醇、n,n-二甲基甲酰胺,二甲基亚砜中的一种或几种。强碱可以是氢化钠、氢化锂、氢化钙、叔丁基锂、甲醇钠、乙醇钠、叔丁醇钠等,反应温度为0~120℃。
[0024]
第3步,化合物5在溶剂中,在酸催化下水解成化合物6,其中溶剂为乙酸乙酯、四氢呋喃、二氯甲烷、乙腈、丙酮等,酸可以为盐酸、硫酸、对甲苯磺酸等,温度为0~40℃。
[0025]
第4步,化合物6在溶剂中,加入自由基引发剂和催化剂,通入空气进行氧化反应得到化合物1。其中引发剂为过氧化苯甲酰,催化剂为n-羟基邻苯二甲酰亚胺且同时加入少量
过渡金属钴、锰或铜的氧化物或盐(醋酸钴、醋酸锰、二氧化锰和碘化亚铜等)。反应溶剂是c3-c6的酮,可以是直链也可以是支链的酮(如丙酮、丁酮、甲基异丁基甲酮),还可以是环己酮或环戊酮等。反应溶剂中也可以加入酯类协同溶剂r1coor2,其中r1可以是c1-c3的直链烷烃,也可以是c2-c3的支链烷烃;r2可以是c2-c4的直链垸烃,也可以是c2-c4的支链烷烃,如乙酸乙酯、乙酸丁酯、丙酸乙酯、醋酸异丙酯等。反应温度是30-80℃。
[0026]
实施例1
[0027]
化合物4的制备:
[0028]
氮气保护下,于反应瓶加入100g化合物1(4-ad),110g原甲酸三乙酯,30g无水乙醇搅拌,降温至0-10℃,加入3g对甲苯磺酸,监控反应完毕。加入50g水搅拌0.5小时,加入8%碳酸钠溶液调节ph至8-9,搅拌2小时,过滤,滤饼烘干得101g化合物4,收率92.1%。
[0029]
化合物6的制备:
[0030]
氮气保护下,于反应瓶加入130g乙基三苯基溴化膦,1000g无水四氢呋喃,搅拌下加入60g乙基三苯基溴化膦,升温至50-60℃反应2小时,滴加22g化合物4(溶于50g四氢呋喃),滴加完毕控温55-60℃反应20小时直至反应完毕。降温加入600g水,搅拌0.5小时,加入500ml乙酸乙酯萃取,重复萃取三次,合并有机层,无水硫酸钠干燥,浓干,加入1000mlthf,加入20ml 5%盐酸,浓干,用正己烷和乙酸乙酯重结晶得到112.8g化合物6,收率91.3%。
[0031]
化合物1的制备:
[0032]
于反应瓶加入50g化合物6,加入400g环己酮和7.5gn-羟基邻苯二甲酰亚胺,搅拌溶清,加入15g过氧化苯甲酰和0.1g二氧化锰,升温至45-50℃,通入空气反应10-15小时,反应完成,浓缩回收环己酮,残留物加入二氯甲烷,过滤,滤液浓缩,甲醇结晶,过滤,烘干得46.5g,收率89.2%。
[0033]
对比例1
[0034]
化合物3至化合物6的制备方法与实施例1相同。
[0035]
化合物1的制备:
[0036]
于反应瓶加入20g化合物6,加入160g环己酮和3g n-羟基邻苯二甲酰亚胺,搅拌溶清,加入6g过氧化苯甲酰,升温至45-50℃,通入空气反应10-15小时,反应完成,浓缩回收环己酮,残留物加入二氯甲烷,过滤,滤液浓缩,甲醇结晶,过滤,烘干得14.3g,收率68.3%。
[0037]
由实施例1和对比例1的对比可知,当催化体系中不加过渡金属锰、钴或铜的氧化物或盐时,催化效果较差。
[0038]
对比例2
[0039]
化合物3至化合物6的制备方法与实施例1相同。
[0040]
化合物1的制备:
[0041]
于反应瓶加入20g化合物6,加入160g环己酮和3g n-羟基邻苯二甲酰亚胺,搅拌溶清,加入6g过氧化苯甲酰和0.1g三氧化铬,升温至45-50℃,通入空气反应10-15小时,反应完成,浓缩回收环己酮,残留物加入二氯甲烷,过滤,滤液浓缩,甲醇结晶,过滤,烘干得10.1g,收率48.2%。
[0042]
由实施例1和对比例1及对比例2的对比可知,当催化体系中加入三氧化铬时,催化效果比不加任何过渡金属的氧化物或盐时还更差。
[0043]
对比例3
[0044]
该对比例为现有技术中由化合物6制备化合物7,再经化合物7制备化合物1的方法。
[0045]
化合物7的制备:
[0046]
于反应瓶加入41g二氧化硒,500ml二氯甲烷,加入160g过氧叔丁醇,搅拌0.5小时,滴加22g化合物6(溶于500ml二氯甲烷),反应完毕,用碳酸氢钠淬灭,二氯甲烷萃取,盐水洗涤,干燥,浓缩,混合物即为化合物7,直接投下一步氧化。
[0047]
化合物1的制备:
[0048]
于反应瓶加入64ml草酰氯,11ml dmso,300ml二氯甲烷,升温45-50℃反应10分钟,滴加化合物7(溶于300ml二氯甲烷),滴加完毕于45-50℃反应1小时,加入三乙胺淬灭,搅拌,降温,再加入二氯甲烷稀释,分别用3n盐酸,10%碳酸氢钠,25%盐水洗涤,无水硫酸钠干燥,正己烷/乙酸乙酯重结晶,得e-古古甾酮(化合物1)17.3g,两步收率75.2%。
[0049]
由实施例1与对比例3的对比可知,实施例1步骤简单,且在同样的反应温度下古古甾酮的收率明显更高。
[0050]
除了实施例1和对比例1、对比例2以外,本发明的发明人还做了如下试验作为补充的实施例和对比例,详见表1。
[0051]
表1
[0052][0053]
表1中,对比例5因为反应不完全,无法后处理,因而无法统计产品收率。此外,在没有特殊指明的情况下,表1中其它实施例和对比例中的其它反应条件均与实施例1相同。
[0054]
由表1可见,实施例2~4中选用醋酸锰、醋酸钴和碘化亚铜代替实施例1中的二氧化锰,所得产品古古甾酮的收率都不如实施例1,但却比对比例1中不添加任何过渡金属的氧化物或盐的催化效果要好。实施例5选用环己酮与乙酸乙酯作为混合有机溶剂,其催化效果也比实施例1略差。对比例4中将本发明的引发剂由过氧化苯甲酰改为使用偶氮二异丁腈,发现其催化效果比实施例1差很多。对比例5~7及对比例1考察了不添加任何过渡金属的氧化物或盐的条件下,反应温度对催化效果的影响,发现反应温度在60-65℃以下时,随着反应温度升高,产品古古甾酮的收率升高;但当反应温度大于65℃时,产品古古甾酮的收率逐步下降。与对比例1相比,对比例8中将有机溶剂由环己酮更换为环戊酮,发现产品古古甾酮的收率略有升高,但升高幅度不大。
[0055]
表2为催化剂当量筛选结果。实施例1中n-羟基邻苯二甲酰亚胺与底物化合物6的质量比为0.15:1,二氧化锰与底物化合物6的质量比为0.002:1,其古古甾酮的收率为89.2%。表2中考察n-羟基邻苯二甲酰亚胺与底物化合物6的不同质量比或者二氧化锰与底物化合物6的不同质量比时的反应结果,见实施例6~11。在没有特殊指明的情况下,实施例6~11中的其它反应条件均与实施例1相同。
[0056]
表2
[0057]
序号实例n-羟基邻苯二甲酰亚胺(w)二氧化锰(w)收率1实施例10.150.00289.2%2实施例60.300.00288.6%3实施例70.0750.00278.9%4实施例80.150.00185.9%5实施例90.150.00588.2%6实施例100.150.0185.6%7实施例110.150.175.9%
[0058]
由表2可见,实施例1和实施例6~7中n-羟基邻苯二甲酰亚胺与式6所示底物的质量比不同,结论是引发剂n-羟基邻苯二甲酰亚胺与式6所示底物的质量比为0.075~0.3:1时均有不错的催化效果,但优选二者的比例为0.1~0.2:1,更优选0.15:1。实施例1和实施例8~11中二氧化锰与式6所示底物的质量比不同,结论是二氧化锰与式6所示底物的质量比为0.001~0.1:1时均有不错的催化效果,但优选二者的比例为0.0015~0.005:1,更优选0.002~0.005:1。
[0059]
上述实施例仅为清楚地说明本发明技术方案所作的举例,而并非是对本发明的实施方式的限定。在不改变本发明基本构思和实质的情况下,任何其它等同技术特征的变换或修改,都应属于本发明权利要求的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1