一种手性苄基硅烷类化合物的制备方法
1.本发明涉及一种手性苄基硅烷类化合物的制备方法,属于有机化合物制备技术领域。
背景技术:
2.有机硅化合物是一类非常重要的试剂、中间体和药物活性分子,由于c-si键容易转化为c-o、c-n和c-c键,所以被应用于许多重要化合物、天然产物和药物的合成及生产中,同时,具有药理活性的有机硅分子也是一类具有发展前途的药物前体。苄基硅烷类化合物属于有机硅化合物,同样在许多重要化合物、天然产物和药物的合成与生产中起到举足轻重的作用。
3.目前,苄基硅烷类化合物的合成方法主要包括:1、炔基硅烷硼氢化后与芳基碘偶联而得到硅基取代苯乙烯类化合物,再在铱催化不对称氢化条件下转变为苄基硅烷类化合物(adv.syn.&cat.,2017,359,2523;tetra.lett.,2016,57,5666);2、取代苯乙烯在钯催化下与三氯硅烷发生不对称氢硅化反应得到苄基氯硅烷,再与格氏试剂反应而得到(j.org.chem.,1986,51,3772);3、手性苄基季铵盐在铜催化下和硅基硼烷反应、或手性苄基醚在镍催化下和硅基格氏试剂反应而得到(angew.chem.int ed.,2020,59,1577;org.lett.,2021,23,1333);4、芳基取代烯丙基醇的磷酸酯在铜催化下与(二甲基苯基)硅基硼硼酸频哪醇酯发生不对称硅基烯丙基化反应而得到(j.org.chem.,2013,78,5007);5、以对亚甲基苯醌类化合物为底物,铜或碳酸铯催化与(二甲基苯基)硅基硼硼酸频哪醇酯发生1,6-硅基加成反应而得到非光活性的苄基硅烷类化合物(chem.commun.,2015,51,17684;cn113429432a;rsc advances,2021,11,17860)。上述已有的方法中除最后一个方法外,都需要过渡金属催化剂,而最后一个方法合成的是非光活性苄基硅烷类化合物;由此看出,目前手性苄基硅烷类化合物合成所采用的方法存在贵重金属的消耗和污染、严格的无水条件和繁琐的操作等问题。因此,发展一种无过渡金属条件下合成手性苄基硅烷类化合物的制备方法既具有重要的科学意义,又具有良好的应用潜力。
4.对亚甲基苯醌类化合物(p-qms),因其独特的骨架结构和特殊的亲电活性,具有更容易发生亲核加成-芳构化反应而构建一系列不同结构、不同类型化合物的性能,使得此类反应底物受到科研工作者们的青睐,在有机合成领域有着举足轻重的地位;同时,以对亚甲基苯醌类化合物为底物,所得到的手性苄基硅烷类化合物通过c-si键转化为c-o、c-n和c-c键,可以被应用于许多重要化合物、天然产物和药物的合成及生产中。目前现有技术中未有以对亚甲基苯醌类化合物为底物,通过无过渡金属催化的不对称1,6-硅基加成反应方法来制备手性苄基硅烷类化合物的报道。为此,提出本发明。
技术实现要素:
5.针对现有技术的不足,本发明提供了一种手性苄基硅烷类化合物的制备方法。本发明的制备方法,所使用的原料廉价、易得,合成操作步骤简单温和,不需要过渡金属催化,
而且能够获得高收率、高化学纯度和高光学纯度的手性苄基硅烷类化合物,适合手性苄基硅烷类化合物的大量制备和规模化生产。
6.本发明的技术方案如下:
7.一种手性苄基硅烷类化合物的制备方法,包括步骤如下:
8.(1)将手性1,2,4-三氮唑盐、碱和溶剂a混合,搅拌反应后得到手性氮杂环卡宾催化剂体系;
9.(2)于溶剂b中,在手性氮杂环卡宾催化剂体系催化下,对亚甲基苯醌类化合物i和(二甲基苯基)硅基硼酸频哪醇酯ii经1,6-硅基加成反应得到手性苄基硅烷类化合物iii;
[0010][0011]
式i化合物结构式中,r1、r2各自独立的选自烷基、环烷基、芳香基、杂环基、烷氧基、芳氧基、卤素、烷氨基或芳氨基;r3为烷基、环烷基、芳香基或杂环基;
[0012]
式iii化合物结构式中,取代基r1、r2、r3与式i化合物结构式中相同。
[0013]
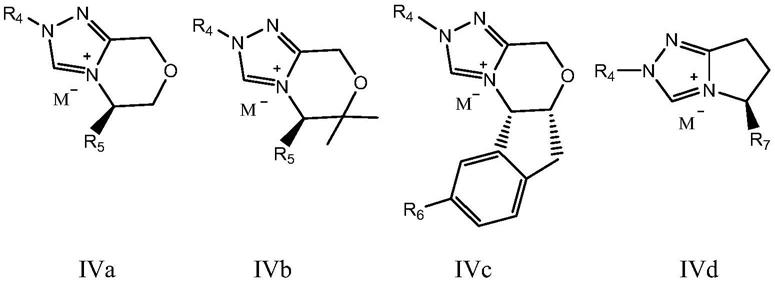
根据本发明优选的,步骤(1)中所述手性1,2,4-三氮唑盐为下述通式iva~ivd所示化合物中的一种:
[0014][0015]
其中,r4、r5、r7各自独立的选自烷基、环烷基、芳香基或杂环基;进一步优选的,r4、r5、r7各自独立的选自c1~c20的烷基、c3~c20的环烷基,c6~c30的芳香基或c2~c30的杂环基;r6为氢、烷基、环烷基、芳香基、杂环基、卤素、硝基、烷氧基或腈基,进一步优选的,r6为氢、c1~c20的烷基、c3~c20的环烷基,c6~c30的芳香基、c2~c30的杂环基、卤素、硝基、烷氧基或腈基;m为bf4、otf或cl;
[0016]
更优选的,所述手性1,2,4-三氮唑盐为下述通式所示化合物中的一种:
[0017][0018]
根据本发明优选的,步骤(1)中所述碱为氨基金属盐、烷氧金属盐、金属氢氧化物、碳酸盐、羧酸盐、硅酸盐或氟化盐;进一步优选的,所述碱为二异丙基氨基锂、叔丁醇钾、氢氧化钾、碳酸铯、碳酸钾、三甲基硅酸钾、醋酸钾、氟化钾或四丁基氟化铵;更优选的,所述碱为碳酸铯、碳酸钾、三甲基硅酸钾、醋酸钾或氟化钾。
[0019]
根据本发明优选的,步骤(1)中所述溶剂a为四氢呋喃、二噁烷或甲基四氢呋喃,进一步优选为四氢呋喃。
[0020]
根据本发明优选的,步骤(1)中所述手性1,2,4-三氮唑盐与碱的摩尔比为1:1~100,进一步优选为1:10~50,更优选为1:20~40。
[0021]
根据本发明优选的,步骤(1)所述手性1,2,4-三氮唑盐的质量与溶剂a的体积之比为0.005~250.0mg:1ml,进一步优选为0.05~25.0mg:1ml,更优选为0.15~5.0mg:1ml。
[0022]
根据本发明优选的,步骤(1)所述反应时间为0.5~12小时。
[0023]
根据本发明优选的,步骤(2)中所述溶剂b为醇类溶剂;进一步优选的,所述溶剂b为甲醇、乙醇、丙醇、异丙醇、三氟乙醇、六氟异丙醇或丁醇;更优选的,所述溶剂b为甲醇、乙醇或三氟乙醇。
[0024]
根据本发明优选的,步骤(2)中所述r1、r2各自独立的选自c1-c20的烷基、c3-c20的环烷基、c6-c30的芳香基、c2-c30的杂环基、c1-c20的烷氧基、c6-c30的芳氧基、c1-c20的烷氨基、c6-c30的芳氨基;进一步优选的,所述r1、r2各自独立的选自叔丁基、甲基、乙基、异丙基、环丙基、环戊基、环己基、苯基、萘基、蒽基、菲基、呋喃基、噻吩基、吡咯基、吲哚基、甲氧基、乙氧基、叔丁氧基、苄氧基、苯氧基、萘氧基、蒽氧基、菲氧基、甲氨基、二甲氨基、乙氨基、二乙氨基、苯氨基、二苯氨基、萘氨基、蒽氨基、菲氨基;所述r3为c1-c20的烷基、c3-c20的环烷基、c6-c30的芳香基、c2-c30的杂环基;进一步优选的,所述r3为甲基、乙基、丙基、异丙基、叔丁基、环丙基、环戊基、环己基、苯基、取代苯基、萘基、蒽基、菲基、呋喃基、噻吩基、吡咯基、吲哚基、吡啶基,所述取代苯基的取代基为甲基、甲氧基、苄氧基、苯氧基、卤素、氰基、酰基、羧基、氨基、硝基或三氟甲基。
[0025]
根据本发明优选的,步骤(2)中所述对亚甲基苯醌类化合物i和手性1,2,4-三氮唑盐的摩尔比为1:0.00001~0.1,进一步优选为1:0.0001~0.05,更优选为1:0.001~0.01。
[0026]
根据本发明优选的,步骤(2)中所述对亚甲基苯醌类化合物i和(二甲基苯基)硅基硼酸频哪醇酯ii的摩尔比为1:1~4.0,进一步优选为1:1~2.0,更优选为1:1~1.5。
[0027]
根据本发明优选的,步骤(2)中,所述1,6-硅基加成反应步骤具体为:将对亚甲基苯醌类化合物i和(二甲基苯基)硅基硼酸频哪醇酯ii加入到手性氮杂环卡宾催化剂体系中,再加入溶剂b得混合液,之后进行反应;混合液中对亚甲基苯醌类化合物i的质量浓度为0.005~0.5g/ml,进一步优选为0.05~0.3g/ml,更优选为0.06~0.3g/ml。
[0028]
根据本发明优选的,步骤(2)中所述1,6-硅基加成反应温度为-35~70℃,进一步优选为0~50℃,更优选为25~40℃。
[0029]
根据本发明优选的,步骤(2)中所述1,6-硅基加成反应时间为0.05~30小时;通过薄层色谱跟踪检测反应进程,对亚甲基苯醌类化合物i消耗完毕,即为反应终点。
[0030]
根据本发明优选的,步骤(2)中所述对亚甲基苯醌类化合物i和(二甲基苯基)硅基硼酸频哪醇酯ii反应后所得反应液的后处理步骤如下:向所得反应液中加入乙酸,所述乙酸与对亚甲基苯醌类化合物i的摩尔比为1:0.1~10,再经蒸馏或减压旋蒸回收溶剂后,得粗产物,将所得粗产物经柱层析分离提纯,得到苄基硼酸酯类化合物;
[0031]
进一步优选的,所述减压旋蒸条件为:旋蒸压力为-0.005~-0.095mpa,旋蒸温度为25~120℃;更优选的,所述旋蒸压力为-0.030~-0.090mpa,旋蒸温度为35~100℃;最优选的,所述旋蒸压力为-0.050~-0.090mpa,旋蒸温度为45~65℃;
[0032]
进一步优选的,所述柱层析分离的洗脱剂为石油醚和三氯甲烷的混合溶剂,所述混合溶剂中石油醚和三氯甲烷的体积比为10:0.1~10,进一步优选为10:1~5,更优选为10:2~4。
[0033]
本发明的合成路线如下:
[0034][0035]
上式中,取代基r1、r2、r3如上述所述。
[0036]
本发明的反应机理如下:
[0037][0038]
手性1,2,4-三氮唑盐和碱在溶剂a中反应生成氮杂环卡宾1,氮杂环卡宾1和(二甲基苯基)硅基硼酸频哪醇酯在含醇的溶剂中反应生成卡宾配合物2(氮杂环卡宾可以和与醇部分交换的(二甲基苯基)硅基硼酸频哪醇酯配位),配合物2作为亲核试剂进攻缺电子的对亚甲基苯醌发生1,6-硅基加成反应得到中间产物3,中间产物3经质子化得到产物手性苄基硅烷类化合物和卡宾硼酸酯配合物4,卡宾硼酸酯配合物4脱去硼酸酯副产物而再生催化剂氮杂环卡宾1。
[0039]
本发明的技术特点及有益效果如下:
[0040]
1、本发明的制备方法仅使用少量的(0.1%)的催化剂手性1,2,4-三氮唑盐,即可催化对亚甲基苯醌类化合物和(二甲基苯基)硅基硼酸频哪醇酯反应得到手性苄基硅烷类化合物,不需要过渡金属,也不需要无水无氧操作;原料种类少,成本低,所使用的原料廉价、易得;制备操作步骤简单温和,无重金属和废水污染,而且能够获得很高的收率和化学纯度以及优异的光学纯度,适合手性苄基硅烷类化合物的大量制备和规模化生产。
[0041]
2、采用本发明的合成方法制备手性苄基硅烷类化合物,能够获得很高的收率和化学纯度及优异的光学纯度;苄基硅烷类化合物的收率最高可达到97%,纯度大于98%,光学纯度可大于95%ee。
具体实施方式
[0042]
下面结合具体实施例对本发明做进一步的说明,但不限于此。
[0043]
同时下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0044]
实施例中所用(二甲基苯基)硅基硼酸频哪醇酯天津希恩思奥普德科技有限公司有售。
[0045]
实施例中所用手性1,2,4-三氮唑盐(s,s
p
)-11按照文献org.biomol.chem.,2015,13,10691报道的方法合成,其结构式如下所示:
[0046][0047]
实施例中所述收率为摩尔收率。
[0048]
实施例1
[0049]
一种手性苄基硅烷类化合物的制备方法,包括步骤如下:
[0050]
(1)向装有磁力搅拌装置的10毫升试管中加入手性1,2,4-三氮唑盐(s,s
p
)-11 4.3毫克(1.0%,0.01mmol)、碳酸铯97.8毫克(30.0%,0.3mmol)和2.4毫升四氢呋喃,室温(25℃)搅拌2小时,得到手性氮杂环卡宾催化剂体系。
[0051]
(2)向上述试管中,加入(二甲基苯基)硅基硼酸频哪醇酯393.3毫克(1.5mmol)、2,6-二叔丁基-4-苯亚甲基-2,5-环己二烯-1-酮ia 294.4毫克(1.0mmol),再加入甲醇2.4毫升,室温(25℃)搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基-4-苯亚甲基-2,5-环己二烯-1-酮完全消耗,停止反应(需2.5小时);向反应液中加入乙酸60毫克(1.0mmol),减压旋蒸以除去溶剂,得粗产物的油状物,所述旋蒸条件为:旋蒸压力为-0.090mpa,旋蒸温度为40-60℃。
[0052]
(3)将所得粗产物经柱层析分离的方法进行提纯,洗脱剂为石油醚和三氯甲烷的混合溶剂,其中石油醚和三氯甲烷的体积比为10:2,可以得到光活性的苄基硅烷类化合物iiia的纯品414mg(收率为96%,纯度为98.8%)。
[0053]
[0054]
所得手性苄基硅烷类化合物iiia的表征数据如下:
[0055]
产物为无色油状物,比旋光度为:[α]
d25
=+5.6(c 0.1,三氯甲烷),经高效液相色谱手性柱分析产物的光学纯度89.1%ee;所述hplc分析条件为:lux cellulose-3柱,乙腈:水:乙酸=60:40:0.1,流速为1.0ml/min,检测器波长为254nm,主峰保留时间为29.1min,对映体的保留时间为26.7min。
[0056]
核磁分析为:1h nmr(500mhz,cdcl3,rt)δ7.41-7.36(m,1h)7.34-7.25(m,6h),7.22-7.15(m,3h),6.92(s,2h),3.69(s,1h),1.40(s,18h),0.34(s,3h),0.32(s,3h)。
[0057]
相似的手性1,2,4-三氮唑盐在上述相同条件下催化(二甲基苯基)硅基硼酸频哪醇酯与2,6-二叔丁基-4-苯亚甲基-2,5-环己二烯-1-酮的不对称1,6-硅基加成反应结果如下表1所示:
[0058]
表1不同手性1,2,4-三氮唑盐催化反应结果
[0059][0060]
根据表中的数据可以看出,手性1,2,4-三氮唑盐连接有缺电子的芳基如五氟苯基时,不对称催化反应进行的很快,但对映选择性并不一定好;如配体(s)-4和(s)-6,尤其是配体(s)-6,可以使反应在5min内完成,反应的对映选择性在70%ee左右,可能原因是:手性1,2,4-三氮唑盐中含有吸电子基团既对卡宾的形成和稳定有利,也可以和与醇部分交换的(二甲基苯基)硅基硼酸频哪醇酯配位、促进对亚甲基苯醌发生1,6-硅基加成反应,还对卡宾硼酸酯配合物脱去硼酸酯副产物而再生催化剂氮杂环卡宾起到了促进的作用,故而这两个手性1,2,4-三氮唑盐的反应活性高。通过对比可以发现,手性1,2,4-三氮唑盐(s,sp)-11的催化性能最好,不但催化反应的活性高,而且反应的对映选择性也最好。
[0061]
实施例2
[0062]
一种手性苄基硅烷类化合物的制备方法,包括步骤如下:
[0063]
(1)向装有磁力搅拌装置的100毫升烧瓶中加入手性1,2,4-三氮唑盐(s,s
p
)-11 4.3毫克(0.1%,0.01mmol)、碳酸铯97.8毫克(3.0%,0.3mmol)和24.0毫升四氢呋喃,室温搅拌2小时,得到手性氮杂环卡宾催化剂体系。
[0064]
(2)向上述试管中,加入(二甲基苯基)硅基硼酸频哪醇酯3.93克(15.0mmol)、2,6-二叔丁基-4-苯亚甲基-2,5-环己二烯-1-酮2.94克(10.0mmol),再加入甲醇24毫升,室温
(25℃)搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基-4-苯亚甲基-2,5-环己二烯-1-酮完全消耗,停止反应(需15.0小时);向反应液中加入乙酸60毫克(1.0mmol),减压旋蒸以除去溶剂,得粗产物的油状物,所述旋蒸条件为:旋蒸压力为-0.090mpa,旋蒸温度为40-60℃。
[0065]
(3)所得粗产物经柱层析分离的方法进行提纯,洗脱剂为石油醚和三氯甲烷的混合溶剂,石油醚和三氯甲烷的体积比为10:2,可以得到光活性的苄基硅烷类化合物的纯品4.05g(收率为94%,纯度为98.6%,光学纯度为86.4%ee)。
[0066]
实施例3
[0067]
一种手性苄基硅烷类化合物的制备方法,包括步骤如下:
[0068]
(1)向装有磁力搅拌装置的10毫升试管中加入手性1,2,4-三氮唑盐(s,s
p
)-11 4.3毫克(1.0%,0.01mmol)、碳酸铯97.8毫克(30.0%,0.3mmol)和2.4毫升四氢呋喃,室温搅拌2小时,得到手性氮杂环卡宾催化剂体系。
[0069]
(2)向上述试管中,加入(二甲基苯基)硅基硼酸频哪醇酯393.3毫克(1.5mmol)、2,6-二叔丁基-4-(4-甲基苯基)亚甲基-2,5-环己二烯-1-酮ib 308.5毫克(1.0mmol),再加入甲醇2.4毫升,室温(25℃)搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基-4-(4-甲基苯基)亚甲基-2,5-环己二烯-1-酮完全消耗,停止反应(需3.0小时);向反应液中加入乙酸60毫克(1.0mmol),减压旋蒸以除去溶剂,得粗产物的油状物,所述旋蒸条件为:旋蒸压力为-0.090mpa,旋蒸温度为40-60℃。
[0070][0071]
(3)将所得粗产物经柱层析分离的方法进行提纯,洗脱剂为石油醚和三氯甲烷的混合溶剂,其中石油醚和三氯甲烷的体积比为10:2,可以得到光活性的苄基硅烷类化合物iiib的纯品419mg(收率为94%,纯度为98.9%)。
[0072]
所得手性苄基硅烷类化合物iiib的表征数据如下:
[0073]
产物为无色油状物,比旋光度为:[α]
d25
=+12.6(c 0.1,三氯甲烷),经高效液相色谱手性柱分析产物的光学纯度92.3%ee;所述hplc分析条件为:lux cellulose-3柱,乙腈:水:乙酸=60:40:0.1,流速为1.0ml/min,检测器波长为254nm,主峰保留时间为29.6min,对映体的保留时间为38.2min。
[0074]
核磁分析为:1h nmr(500mhz,cdcl3,rt)δ7.40-7.36(m,1h)7.35-7.31(m,4h),7.13-7.07(m,4h),6.92(s,2h),4.98(s,1h),3.65(s,1h),2.35(s,3h),1.40(s,18h),0.34(s,3h),0.31(s,3h)。
[0075]
实施例4
[0076]
一种手性苄基硅烷类化合物的制备方法,包括步骤如下:
[0077]
(1)向装有磁力搅拌装置的10毫升试管中加入手性1,2,4-三氮唑盐(s,s
p
)-11 4.3毫克(1.0%,0.01mmol)、碳酸铯97.8毫克(30.0%,0.3mmol)和2.4毫升四氢呋喃,室温搅拌2小时,得到手性氮杂环卡宾催化剂体系。
[0078]
(2)向上述试管中,加入(二甲基苯基)硅基硼酸频哪醇酯393.3毫克(1.5mmol)、2,6-二叔丁基-4-(4-氯苯基)亚甲基-2,5-环己二烯-1-酮ic 328.9毫克(1.0mmol),再加入甲醇2.4毫升,室温(25℃)搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基-4-(4-氯苯基)亚甲基-2,5-环己二烯-1-酮完全消耗,停止反应(需4.0小时);向反应液中加入乙酸60毫克(1.0mmol),减压旋蒸以除去溶剂,得粗产物的油状物,所述旋蒸条件为:旋蒸压力为-0.090mpa,旋蒸温度为40-60℃。
[0079][0080]
(3)将所得粗产物经柱层析分离的方法进行提纯,洗脱剂为石油醚和三氯甲烷的混合溶剂,其中石油醚和三氯甲烷的体积比为10:3,可以得到光活性的苄基硅烷类化合物的纯品iiic 428mg(收率为92%,纯度为98.8%)。
[0081]
所得手性苄基硅烷类化合物iiic的表征数据如下:
[0082]
产物为无色油状物,比旋光度为:[α]
d25
=+5.3(c 0.1,三氯甲烷),经高效液相色谱手性柱分析产物的光学纯度93.2%ee;所述hplc分析条件为:lux cellulose-3柱,乙腈:水:乙酸=70:30:0.1,流速为1.0ml/min,检测器波长为254nm,主峰保留时间为20.7min,对映体的保留时间为25.3min。
[0083]
核磁分析为:1h nmr(500mhz,cdcl3,rt)δ7.41-7.36(m,1h)7.34-7.26(m,4h),7.24-7.21(m,2h),7.11-7.08(m,2h),6.89(s,2h),5.02(s,1h),3.65(s,1h),1.39(s,18h),0.34(s,3h),0.32(s,3h)。
[0084]
实施例5
[0085]
一种手性苄基硅烷类化合物的制备方法,包括步骤如下:
[0086]
(1)向装有磁力搅拌装置的10毫升试管中,加入手性1,2,4-三氮唑盐(s,s
p
)-11 4.3毫克(1.0%,0.01mmol)、碳酸铯97.8毫克(30.0%,0.3mmol)和2.4毫升四氢呋喃,室温搅拌2小时,得到手性氮杂环卡宾催化剂体系。
[0087]
(2)向上述试管中,加入(二甲基苯基)硅基硼酸频哪醇酯393.3毫克(1.5mmol)、2,6-二叔丁基-4-(2-萘基)亚甲基-2,5-环己二烯-1-酮id 344.5毫克(1.0mmol),再加入甲醇2.4毫升,室温(25℃)搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基-4-(2-萘基)亚甲基-2,5-环己二烯-1-酮完全消耗,停止反应(需0.5小时);向反应液中加入乙酸
60毫克(1.0mmol),减压旋蒸以除去溶剂,得粗产物的油状物,所述旋蒸条件为:旋蒸压力为-0.090mpa,旋蒸温度为40-60℃。
[0088][0089]
(3)将所得粗产物经柱层析分离的方法进行提纯,洗脱剂为石油醚和三氯甲烷的混合溶剂,其中石油醚和三氯甲烷的体积比为10:2,可以得到光活性的苄基硅烷类化合物的纯品iiid 448mg(收率为93%,纯度为98.8%)。
[0090]
所得手性苄基硅烷类化合物iiid的表征数据如下:
[0091]
产物为无色油状物,比旋光度为:[α]
d25
=+15.5(c 0.1,三氯甲烷),经高效液相色谱手性柱分析产物的光学纯度95.1%ee;所述hplc分析条件为:lux cellulose-3柱,乙腈:水:乙酸=70:30:0.1,流速为1.0ml/min,检测器波长为254nm,主峰保留时间为13.4min,对映体的保留时间为35.6min。
[0092]
核磁分析为:1h nmr(500mhz,cdcl3,rt)δ7.83(d,j=7.8hz,1h),7.76-7.72(m,2h),7.65(s,1h),7.48-7.38(m,3h),7.36-7.31(m,5h),7.01(s,2h),3.88(s,1h),1.41(s,18h),0.38(s,3h),0.35(s,3h)。
[0093]
实施例6
[0094]
一种手性苄基硅烷类化合物的制备方法,包括步骤如下:
[0095]
(1)装有磁力搅拌装置的10毫升试管中,加入手性1,2,4-三氮唑盐(s,s
p
)-11 4.3毫克(1.0%,0.01mmol)、碳酸铯97.8毫克(30.0%,0.3mmol)和2.4毫升四氢呋喃,室温搅拌2小时,得到手性氮杂环卡宾催化剂体系。
[0096]
(2)向上述试管中,加入(二甲基苯基)硅基硼酸频哪醇酯393.3毫克(1.5mmol)、2,6-二叔丁基-4-(2-噻吩基)亚甲基-2,5-环己二烯-1-酮ie 300.5毫克(1.0mmol),再加入甲醇2.4毫升,室温(25℃)搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基-4-(2-噻吩基)亚甲基-2,5-环己二烯-1-酮完全消耗,停止反应(需4.0小时);向反应液中加入乙酸60毫克(1.0mmol),减压旋蒸以除去溶剂,得粗产物的油状物,所述旋蒸条件为:旋蒸压力为-0.090mpa,旋蒸温度为40-60℃。
[0097][0098]
(3)将所得粗产物经柱层析分离的方法进行提纯,洗脱剂为石油醚和三氯甲烷的混合溶剂,其中石油醚和三氯甲烷的体积比为10:2,可以得到光活性的苄基硅烷类化合物iiie的纯品393mg(收率为90%,纯度为98.2%)。
[0099]
所得手性苄基硅烷类化合物iiie的表征数据如下:
[0100]
产物为无色油状物,比旋光度为:[α]
d25
=-6.2(c 0.1,三氯甲烷),经高效液相色谱手性柱分析产物的光学纯度86.3%ee;所述hplc分析条件为:lux cellulose-3柱,乙腈:水:乙酸=60:40:0.1,流速为1.0ml/min,检测器波长为254nm,主峰保留时间为27.1min,对映体的保留时间为24.4min。
[0101]
核磁分析为:1h nmr(500mhz,cdcl3,rt)δ7.40-7.36(m,1h)7.33-7.26(m,4h),7.09-7.05(m,1h),6.94-6.91(m,1h),6.89(s,2h),6.81-6.76(m,1h),4.98(s,1h),3.93(s,1h),1.38(s,18h),0.34(s,3h),0.33(s,3h)。
[0102]
实施例7
[0103]
一种手性苄基硅烷类化合物的制备方法,包括步骤如下:
[0104]
(1)向装有磁力搅拌装置的10毫升试管中加入手性1,2,4-三氮唑盐(s,s
p
)-11 4.3毫克(1.0%,0.01mmol)、碳酸铯97.8毫克(30.0%,0.3mmol)和2.4毫升四氢呋喃,室温搅拌2小时,得到手性氮杂环卡宾催化剂体系。
[0105]
(2)向上述试管中,加入(二甲基苯基)硅基硼酸频哪醇酯393.3毫克(1.5mmol)、2,6-二叔丁基-4-亚乙基-2,5-环己二烯-1-酮if 232.4毫克(1.0mmol),再加入甲醇2.4毫升,室温(25℃)搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基-4-亚乙基-2,5-环己二烯-1-酮完全消耗,停止反应(需2.5小时);向反应液中加入乙酸60毫克(1.0mmol),减压旋蒸以除去溶剂,得粗产物的油状物,所述旋蒸条件为:旋蒸压力为-0.090mpa,旋蒸温度为40-60℃。
[0106][0107]
(3)将所得粗产物经柱层析分离的方法进行提纯,洗脱剂为石油醚和三氯甲烷的
混合溶剂,其中,石油醚和三氯甲烷的体积比为10:3,可以得到光活性的苄基硅烷类化合物iiif的纯品317mg(收率为86%,纯度为98.1%)。
[0108]
所得手性苄基硅烷类化合物iiif的表征数据如下:
[0109]
产物为无色油状物,比旋光度为:[α]
d25
=+10.6(c 0.1,三氯甲烷),经高效液相色谱手性柱分析产物的光学纯度78.4%ee;所述hplc分析条件为:lux cellulose-3柱,乙腈:水:乙酸=70:30:0.1,流速为1.0ml/min,检测器波长为254nm,主峰保留时间为32.8min,对映体的保留时间为26.2min。
[0110]
核磁分析为:1h nmr(500mhz,cdcl3,rt)δ7.41-7.36(m,1h)7.36-7.32(m,4h),6.69(s,2h),2.34-2.29(m,1h),1.40(s,18h),1.37(d,j=7.55hz,3h),0.27(s,3h),0.24(s,3h)。
[0111]
实施例8-13
[0112]
一种手性苄基硅烷类化合物的制备方法,包括步骤如下:
[0113]
(1)向装有磁力搅拌装置的10毫升试管中加入手性1,2,4-三氮唑盐(s,s
p
)-11 4.3毫克(1.0%,0.01mmol)、碱(种类和用量如下表2所示)和2.4毫升四氢呋喃,室温搅拌2小时,得到手性氮杂环卡宾催化剂体系。
[0114]
(2)向上述试管中,加入(二甲基苯基)硅基硼酸频哪醇酯393.3毫克(1.5mmol)、2,6-二叔丁基-4-苯亚甲基-2,5-环己二烯-1-酮ia 294.4毫克(1.0mmol),再加入甲醇2.4毫升,室温(25℃)搅拌反应至表2中指定时间;向反应液中加入乙酸60毫克(1.0mmol),减压旋蒸以除去溶剂,得粗产物的油状物,所述旋蒸条件为:旋蒸压力为-0.090mpa,旋蒸温度为40-60℃。
[0115]
(3)将所得粗产物经柱层析分离的方法进行提纯,洗脱剂为石油醚和三氯甲烷的混合溶剂,其中石油醚和三氯甲烷的体积比为10:2,可以得到光活性的苄基硅烷类化合物的纯品,收率和不对称选择性见下表2。
[0116]
表2
[0117][0118]
对比例1
[0119]
一种手性苄基硅烷类化合物的制备方法如实施例1所述,所不同的是:手性1,2,4-三氮唑盐为(s)-12,结构式如下所示。
[0120][0121]
本对比例中使用手性1,2,4-三氮唑盐(s)-12为催化剂,所得产物的收率为85%,但是其光学纯度为0%ee,为消旋产物,得不到手性苄基硅烷类化合物。
[0122]
对比例2-10
[0123]
一种手性苄基硅烷类化合物的制备方法,包括步骤如下:
[0124]
(1)向装有磁力搅拌装置的10毫升试管中加入手性1,2,4-三氮唑盐(s,s
p
)-11 4.3毫克(1.0%,0.01mmol)、碱(种类和量如下表3所示)和2.4毫升溶剂a,室温搅拌2小时,得到手性氮杂环卡宾催化剂体系。
[0125]
(2)向上述试管中,加入(二甲基苯基)硅基硼酸频哪醇酯393.3毫克(1.5mmol)、2,6-二叔丁基-4-苯亚甲基-2,5-环己二烯-1-酮ia 294.4毫克(1.0mmol),再加入溶剂b 2.4毫升,室温(25℃)搅拌反应至指定时间(反应时间见下表3);向反应液中加入乙酸60毫克(1.0mmol),减压旋蒸以除去溶剂,得粗产物的油状物,所述旋蒸条件为:旋蒸压力为-0.090mpa,旋蒸温度为40-60℃。
[0126]
(3)将所得粗产物经柱层析分离的方法进行提纯,洗脱剂为石油醚和三氯甲烷的混合溶剂,石油醚和三氯甲烷的体积比为10:2,可以得到苄基硅烷类化合物的纯品(收率和不对称选择性见下表3)。
[0127]
表3
[0128][0129]
从表3中可以看出,碱以及溶剂的种类对于产物的收率以及光学纯度具有较大的影响。当碱为三乙胺或dbu时,所得产物的收率虽然较高,但是所得产物为消旋产物,得不到
手性苄基硅烷类化合物;并且本发明需要在四氢呋喃、二噁烷或甲基四氢呋喃和醇类溶剂的混合体系中,才能得到较高收率和光学纯度的手性苄基硅烷类化合物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1