一种制备愈创薁吲哚化合物的方法
1.本发明属于化合物制备领域,具体涉及一种制备愈创薁吲哚化合物的方法。
背景技术:
2.愈创薁(guaiazulene),又称愈创蓝油烃或愈创蓝油烃薁,化学系统命名为1,4
‑
二甲基
‑7‑
异丙基薁。其化学结构式由七元环和五元环稠合而成,与萘为同分异构体,符合休克尔规则,具有芳香性,五元环上1,3
‑
位置倾向于带负电,较容易进行亲电性取代反应,而七元环上的4,6,8
‑
位置倾向带正电荷,较容易进行亲核性反应,因此,愈创薁拥有较高的化学活性。
3.通过愈创薁结构简单改性获得的分子薁磺酸钠,又称呱仑酸钠,其特有的抗炎、抗癌和抗溃疡等特性,已在临床应用于眼睛和牙周膜炎的治疗以及心血管疾病等。另外,在化妆品领域,愈创薁及其衍生物也广泛应用于具有消炎、润肤、保湿和抗皱等功能的乳液中。在其它领域,包括可用作染料着色剂,激光打印和静电复印;还可用作滤光片,液晶显示器,可再充电电池,荧光转化膜,光学记录器和光感受器等。因此能够较为简易、高效对愈创薁结构改性获得结构类型多样的化合物显得尤为重要。
4.基于薁核骨架的修饰合成薁类衍生物是目前研究的热点,其优势在于薁核具有较高的化学活性,因此可以简单、高效地嫁接其它基团,获得结构多样的薁类衍生物。含氮杂环化合物广泛存在于自然界中,因其具有多种生物活性,使其广泛存在医药和农药分子中。在材料领域也能见到其身影。因此,将愈创薁基和氮杂环基团二合为一的愈创薁取代氮杂环衍生物预期具有重要的研究价值。
技术实现要素:
5.目前制备愈创薁吲哚化合物的方法大多需要采用贵金属作为催化剂,成本高昂。本发明的目的是解决现有技术的不足,提供一种能够在室温条件下,采用更价廉易得的酸作为催化剂制备愈创薁吲哚化合物的方法,目前还未见过报道酸催化2
‑
吲哚甲醇化合物与薁衍生物的c3位功能化反应。具体采用以下的技术方案:
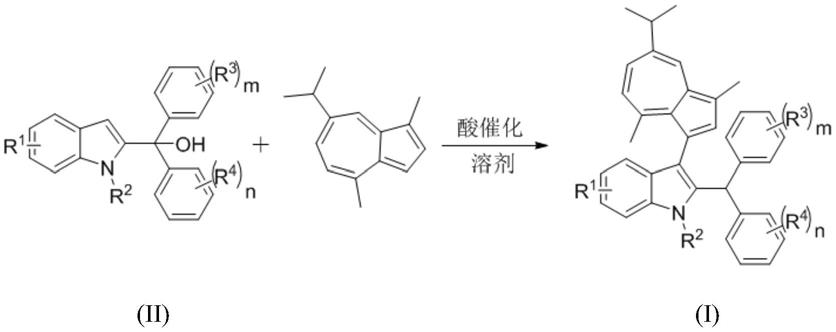
6.一种制备愈创薁吲哚化合物的方法,所述方法是在溶剂中,以愈创薁和通式ii所示化合物为原料,在酸催化剂的条件下反应得到通式i所示化合物,反应过程如下:
[0007][0008]
其中,r1选自h、c1~c6烷基、苯基、卤素、三氟甲基、三氟甲氧基、c1~c4烷氧基;优选为h、甲氧基、卤素;
[0009]
r2选自h、c1~c6烷基;优选为h、甲基;
[0010]
r3选自h、c1~c6烷基、烯基、苯基、卤素、三氟甲基、三氟甲氧基、c1~c4烷氧基,其中m=1~5;进一步地,r3优选为h、甲基、甲氧基、卤素、叔丁基;进一步地,m=1、2、3;
[0011]
r4选自h、c1~c6烷基、烯基、苯基、卤素、三氟甲基、三氟甲氧基、c1~c4烷氧基,其中n=1~5;r4优选为h、甲基、甲氧基、卤素、叔丁基;进一步地,n=1、2、3;
[0012]
所述酸催化剂为布朗斯特酸、路易斯酸和固体酸中的至少一种;所述溶剂为四氢呋喃、乙腈、n,n
‑
二甲基甲酰胺、苯、己烷、氯仿、四氯化碳、丙酮、乙醇、甲醇、甲苯、二氯甲烷、1,2
‑
二氯甲烷、苯甲醚、甲基叔丁基醚、二氧六环、乙二醇二甲醚中的至少1种。
[0013]
在反应中,所述愈创薁的物质的量为通式ii所示化合物的1~3倍。优选为1~1.5倍。所述酸催化剂的物质的量为所述通式ii所示化合物物质的量的0.1%~50%。优选为1%~20%。所述溶剂,其用量满足反应要求即可,优选通式ii所示化合物与溶剂的量之比为1mmol:1~10ml。
[0014]
所述布朗斯特酸为盐酸、硫酸、硝酸、磷酸、乙酸、苯甲酸、甲酸、丙酸、三氟乙酸、三氟甲磺酸、苯磺酸、对甲苯磺酸、对甲苯磺酸一水合物、樟脑磺酸、酒石酸中的至少一种。所述路易斯酸为氯化铁、氯化铜、氯化铝、三氟甲磺酸铜中的至少一种。所述固体酸为酸性离子树脂、杂多酸中的至少一种。
[0015]
所述反应过程中的反应温度为25℃至溶剂沸点温度(也可称为溶剂回流温度),反应时间为10min
‑
24h。优选5h~15h。
[0016]
除另有说明外,本文中使用的术语具有以下含义。
[0017]
本文中使用的术语“烷基”包括直链烷基和支链烷基。如提及单个烷基如“丙基,”则只特指直链烷基,如提及单个支链烷基如“异丙基,”则只特指支链烷基。例如,“c4以下烷基”包括甲基、乙基、正丙基、异丙基、正丁基和叔丁基等。类似的规则也适用于本说明书中使用的其它基团。
[0018]
本文中使用术语“卤素”包括氟、氯、溴、碘。
[0019]
本文中所述c1~c4烷氧基为具有如下结构的基团:
‑
o
‑
m1,其中,m1为c1~c4烷基,如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、叔丁氧基。
[0020]
上述技术方案(r3)
m
中,m=1~5指r3在苯基上的取代可为单取代或多位取代,可为1、2、3、4或5取代。m=1时为单取代,单取代的取代位可为2、3或4位;m=2、3、4或5时,为多位
取代,其中,m=2为双取代,双取代的取代位为2,3
‑
、2,4
‑
、2,5
‑
、2,6
‑
、3,4
‑
、3,5
‑
;m=3为三取代,三取代的取代位为2,3,4
‑
、2,3,5
‑
、2,3,6
‑
、3,4,5
‑
。
[0021]
上述技术方案(r4)
n
中,n=1~5指r4在苯基上的取代可为单取代或多位取代,可为1、2、3、4或5取代。n=1时为单取代,单取代的取代位可为2、3或4位;n=2、3、4或5时,为多位取代,其中,n=2为双取代,双取代的取代位为2,3
‑
、2,4
‑
、2,5
‑
、2,6
‑
、3,4
‑
、3,5
‑
;n=3为三取代,三取代的取代位为2,3,4
‑
、2,3,5
‑
、2,3,6
‑
、3,4,5
‑
。
[0022]
表1中列举了上述反应中各个原料化合物取代基的具体结构。
[0023]
表1
[0024]
[0025]
[0026]
[0027]
[0028]
[0029]
[0030]
[0031][0032]
本发明的有益效果为:本发明提供了一种全新制备愈创薁吲哚化合物的方法,无需贵金属作为催化剂,原料价廉易得,反应过程高效、简便。
附图说明
[0033]
图1所示为化合物1的
13
c
‑
dept和
13
c
‑
apt谱图。
具体实施方式
[0034]
以下将结合实施例和附图对本发明的构思及产生的技术效果进行清楚、完整的描述,以充分地理解本发明的目的、方案和效果。下述实施例中所述试验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0035]
实施例1:
[0036]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物1):
[0037]
室温下,向氮气保护的25ml schlenk瓶中依次加入2
‑
吲哚二苯甲醇90mg(0.3mmol)、1.3倍摩尔量的愈创薁77mg(0.39mmol)、无水氯仿(溶剂)2ml、催化剂(
±
)
‑
10
‑
樟脑磺酸7mg(10mol%),反应体系为蓝色液体,置于25℃反应12小时。后处理通过旋转蒸发仪除去溶剂,通过柱层析获得目标化合物,填充料为硅胶,洗脱剂为石油醚:乙酸乙酯(100:1~20:1),产品为蓝色固体,分离收率98%。
[0038]
实施例2:
[0039]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)
‑5‑
氯吲哚的制备(化合物2):
[0040]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
(5
‑
氯吲哚)二苯甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率98%。
[0041]
实施例3:
[0042]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)
‑5‑
溴吲哚的制备(化合物3):
[0043]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
(5
‑
溴吲哚)二苯甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率89%。
[0044]
实施例4:
[0045]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)
‑5‑
甲氧基吲哚的制备(化合物4):
[0046]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
(5
‑
甲氧吲哚)二苯甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率77%。
[0047]
实施例5:
[0048]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)
‑6‑
氟基吲哚的制备(化合物5):
[0049]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
(6
‑
氟吲哚)二苯甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率98%。
[0050]
实施例6:
[0051]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)
‑6‑
溴基吲哚的制备(化合物6):
[0052]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
(6
‑
溴吲哚)二苯甲醇,然后按
照实施例1同样的方法进行,获得目标化合物分离收率90%。
[0053]
实施例7:
[0054]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)
‑6‑
甲氧基吲哚的制备(化合物7):
[0055]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
(6
‑
甲氧吲哚)二苯甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率82%。
[0056]
实施例8:
[0057]2‑
[二(4
‑
甲基苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物8):
[0058]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
吲哚二(4
‑
甲基苯基)甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率96%。
[0059]
实施例9:
[0060]2‑
[二(4
‑
甲氧基苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物9):
[0061]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
吲哚二(4
‑
甲氧基苯基)甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率94%。
[0062]
实施例10:
[0063]2‑
[二(4
‑
叔丁基苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物10):
[0064]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
吲哚二(4
‑
叔丁基苯基)甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率90%。
[0065]
实施例11:
[0066]2‑
[二(4
‑
氟苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物11):
[0067]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
吲哚二(4
‑
氟苯基)甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率97%。
[0068]
实施例12:
[0069]2‑
[二(4
‑
氯苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物12):
[0070]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
吲哚二(4
‑
氯苯基)甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率96%。
[0071]
实施例13:
[0072]2‑
[二(2
‑
甲基苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物13):
[0073]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
吲哚二(2
‑
甲基苯基)甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率81%。
[0074]
实施例14:
[0075]2‑
[二(2
‑
甲氧基苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物14):
[0076]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
吲哚二(2
‑
甲氧基苯基)甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率98%。
[0077]
实施例15:
[0078]2‑
[二(3
‑
甲基苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物15):
[0079]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
吲哚二(3
‑
甲基苯基)甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率98%。
[0080]
实施例16:
[0081]2‑
[二(3
‑
氯苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物16):
[0082]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
吲哚二(3
‑
氯苯基)甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率95%。
[0083]
实施例17:
[0084]2‑
[二(3
‑
甲氧基苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物17):
[0085]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
吲哚二(3
‑
甲氧基苯基)甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率96%。
[0086]
实施例18:
[0087]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)
‑
n
‑
甲基吲哚的制备(化合物18):
[0088]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
(n
‑
甲基吲哚)二苯基甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率98%。
[0089]
实施例19:
[0090]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)
‑
1,7
‑
二甲基吲哚的制备(化合物19):
[0091]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
(1,7
‑
二甲基吲哚)二苯基甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率97%。
[0092]
实施例20:
[0093]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)
‑
n
‑
甲基
‑4‑
氯吲哚的制备(化合物20):
[0094]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
(n
‑
甲基
‑4‑
氯吲哚)二苯基甲醇,然后按照实施例1同样的方法进行,获得目标化合物分离收率95%。
[0095]
实施例21:
[0096]2‑
[对甲氧二苯基甲基]
‑3‑
(1
‑
愈创薁)
‑
n
‑
甲基吲哚的制备(化合物21):
[0097]
将实施例1中的2
‑
吲哚二苯甲醇换成同摩尔量的2
‑
(n
‑
甲基吲哚)
‑4’‑
甲氧基二苯基甲醇,反应温度设置为70℃,然后按照实施例1同样的方法进行,获得目标化合物分离收率89%。
[0098]
实施例22:
[0099]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物1):
[0100]
将实施例1中的无水氯仿换成无水丙酮,然后按照实施例1同样的方法进行,获得目标化合物液相收率95%(以三苯基膦为内标)。
[0101]
实施例23:
[0102]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物1):
[0103]
将实施例1中的无水氯仿换成无水乙醚,然后按照实施例1同样的方法进行,获得目标化合物液相收率92%(以三苯基膦为内标)。
[0104]
实施例24:
[0105]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物1):
[0106]
将实施例1中的催化剂(
±
)
‑
10
‑
樟脑磺酸换成一水合对甲苯磺酸,然后按照实施例1同样的方法进行,获得目标化合物液相收率89%(以三苯基膦为内标)。
[0107]
实施例25:
[0108]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物1):
[0109]
将实施例1中的催化剂(
±
)
‑
10
‑
樟脑磺酸换成三氟甲磺酸铋,然后按照实施例1同样的方法进行,获得目标化合物液相收率20%(以三苯基膦为内标)。
[0110]
实施例26:
[0111]2‑
[二(苯基)甲基]
‑3‑
(1
‑
愈创薁)吲哚的制备(化合物1):
[0112]
将实施例1中的催化剂(
±
)
‑
10
‑
樟脑磺酸换成15,添加量为10mg,然后按照实施例1同样的方法进行,获得目标化合物液相收率89%(以三苯基膦为内标)。
[0113]
表2所示为合成的具体化合物1
‑
21的结构、物理性质及1h nmr数据,但本发明并不仅限于这些化合物。
[0114]
表2
[0115]
[0116]
[0117]
[0118][0119]
以上所述,只是本发明的较佳实施例而已,本发明并不局限于上述实施方式,只要其以相同的手段达到本发明的技术效果,都应属于本发明的保护范围。在本发明的保护范围内其技术方案和/或实施方式可以有各种不同的修改和变化。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1