一种动态调控磷酸葡糖异构酶产组氨酸的基因工程菌株及其构建方法与应用与流程
1.本发明属于基因工程技术领域,涉及工业微生物的育种,特别涉及一种动态调控磷酸葡糖异构酶生产组氨酸的基因工程菌株及其构建方法与应用。
背景技术:
2.大肠杆菌中的磷酸葡糖异构酶,由pgi基因编码,催化糖酵解途径第二步反应,是调控糖酵解途径与磷酸戊糖途径代谢流量分配的关键节点。磷酸戊糖途径为胞内各种反应过程提供还原力nadph,参与脂肪酸和固醇类物质以及一些重要氨基酸如苏氨酸、缬氨酸、精氨酸等的合成。另外,该途径的中间产物,如5-磷酸-核糖、4-磷酸-赤藓糖等还可以为芳香族氨基酸、核苷等众多胞内重要物质的合成提供原料。因此,降低磷酸葡糖异构酶的表达,以增强磷酸戊糖途径的合成通量是非常重要的改造策略。而糖酵解途径是葡萄糖的主要代谢路径,也是细胞重要的供能途径,与细胞生长关系密切,因此糖酵解途径代谢流量过低会严重影响菌体生长,目标产物的合成也会受到限制。因此,合理调控糖酵解途径和磷酸戊糖途径的代谢流量分配,才能保证在菌体适度生长的前提下,促进磷酸戊糖途径相关产物的合成。
3.目前,在多种产物微生物细胞工厂的构建过程中,都对磷酸葡糖异构酶的表达进行了调控。一种方法是直接敲除磷酸葡糖异构酶的编码基因pgi。发明人在前期构建组氨酸生产菌的过程中曾通过直接敲除磷酸葡糖异构酶的编码基因pgi,增强磷酸戊糖途径的通量,虽然使得组氨酸的产率大幅提升,但是因为pgi基因的敲除大幅降低了糖酵解途径的合成通量,严重妨碍了菌体生长,导致组氨酸的生产强度明显降低(highly efficient production of l-histidine from glucose by metabolically engineered escherichia coli[j].acs synthetic biology,2020,9,7,1813-1822)。温廷益(cn106459886a,cn110117568a)等构建组氨酸工程菌的过程中为了强化磷酸戊糖途径,增强组氨酸合成前体物prpp的供应,敲除pgi基因对菌体的生长有明显的抑制效果且l-组氨酸产量由1.18g/l降低至0.77g/l。单独对磷酸戊糖途径关键酶zwf过表达,产量由1.18g/l升至1.50g/l;敲除pgi基因的同时过表达zwf基因,产量由1.18g/l升至2.40g/l,l-组氨酸产量相较对照菌株产量提升102%。说明增强中心代谢流流向磷酸戊糖途径,缓解了糖代谢压力,协调了糖酵解途径与磷酸戊糖途径的碳流平衡,产量增加。但由于静态改造无法调节基因表达强度,较低的酶活导致菌体生长受限,导致生产强度大幅下降。另外,还有一些案例是通过敲除pgi基因调控中心代谢碳流分配,提高葡萄糖利用率及nadph供给,以促进目标产物的合成(cn1270631a,cn110835621a,cn110656073a,cn105441496a)。这些案例中同样存在菌体生长受阻,降低生产强度的问题。
[0004]
另外一种方法是利用诱导型启动子调控pgi基因表达,可在一定程度上缓解上述生长问题。牛腾飞等人(cn107760643a)通过利用启动子、glms核酶突变体、rbs元件进行组合优化pgi的表达,使重组菌株n3531-g的glcnac产量由14.45g/l增加至18.45g/l,细胞干
重达到15.2g/l,副产物乙偶姻的浓度达到18.2g/l,转化率为0.283g n-乙酰氨基葡萄糖/g葡萄糖;刘龙等人(cn106148260a)利用木糖诱导型启动子调控pgi基因表达,明显提升了乙酰氨基葡萄糖的产量;张杰等人(cn112961792a)通过甘油诱导型启动子调控pgi基因表达,利用甘油和葡萄糖在培养基中的添加时间来控制菌体生长及肌醇的生产,提高了毕赤酵母工程菌株肌醇的生产能力。但是,利用诱导型启动子调控pgi的转录表达同样存在明显的弊端:一是不同诱导型启动子的强度不同,调控的严谨程度也存在差别,因此很难找到合适的启动子;二是诱导型启动子的使用需要把控合适的诱导时机,过早的切换虽然有利于转化率的提升,但易对生长造成较大影响,使菌株无法达到最优产量及产率,而代谢通路的特异性则使得此类方法并不具有普适性;三是诱导物的添加会增加生产成本,并且这种诱导工艺使发酵过程控制变得更为复杂,也容易造成生产的不稳定。
[0005]
综上所述,直接敲除pgi基因虽有助于磷酸戊糖途径相关产物产率的提升,但是菌体生长会受到明显的抑制,从而导致产量或者生产强度的大幅下降,不利于工业化应用。而利用诱导型启动子调控pgi基因的表达时,不同启动子的强度不同,转录的严谨程度也有差异,会影响该方法的使用效果,另外诱导物的添加在增加生产成本的同时也使工艺控制的难度增加,同样不利于产业化应用。
技术实现要素:
[0006]
针对上述问题,本发明的目的是通过一种动态调控磷酸葡糖异构酶表达的方法,可以在满足菌体生长的前提下,有效降低糖酵解途径的通量,加强磷酸戊糖途径,该方法应用于组氨酸工程菌中可以显著提高组氨酸的合成效率。
[0007]
为了实现上述目的,本发明采取如下的技术方案:
[0008]
第一方面,本发明提供了一种大肠杆菌基因工程菌株,该基因工程菌株以大肠杆菌色氨酸操纵子的启动子p
trp
调控pgi基因的转录表达,并且可选的引入以酰基高丝氨酸内酯ahl作为信号分子介导的群体感应系统调控trpr基因的转录表达。
[0009]
第二方面,本发明提供了如上所述的大肠杆菌基因工程菌株的构建方法,包括:在大肠杆菌中,以色氨酸操纵子的启动子p
trp
替换了pgi基因原有启动子;可选的引入ahl合成酶基因esai和群体感应转录调控因子esar的编码基因esar,并且以启动子p
esar-p
替换了trpr基因原有启动子。
[0010]
第三方面,本发明提供了如上所述的大肠杆菌基因工程菌株在高产组氨酸中的用途。
[0011]
本发明的有益效果如下:
[0012]
本发明的关键在于用一种自调控的级联反馈系统动态调控磷酸葡糖异构酶的转录表达,与现有技术相比,本发明所提供的动态调控大肠杆菌磷酸葡糖异构酶的方法,可以在满足菌体生长所需的前提下,有效降低糖酵解途径通量,增强磷酸戊糖途径通量,将该方法应用于组氨酸工程菌的构建,所得菌株摇瓶发酵24h后,相较于对照菌株,组氨酸的产量由3.1g/l提高至6.5g/l,糖酸转化率由3.9%提升至11.7%,产量、转化率和单位菌体产量分别提升109.7%、200%和242.4%,效果显著。
[0013]
本发明中磷酸葡糖异构酶的调控是一种自调控方法,无须诱导物的添加。并且通过调整酰基高丝氨酸内酯(ahl)合成酶编码基因esai的转录强度(如启动子替换)可拓宽对
磷酸葡糖异构酶强度的调控范围。另外,该方法具有较好的普适性,可应用于增强其它磷酸戊糖途径相关产物,如芳香族氨基酸、核苷类等物质的合成。
附图说明
[0014]
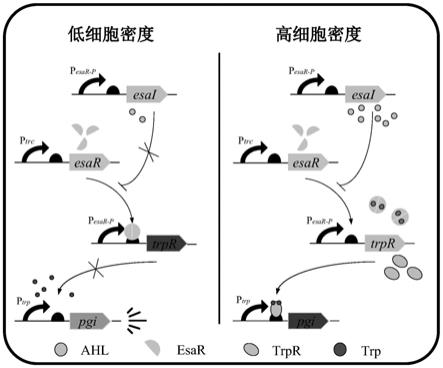
图1:动态调控磷酸葡糖异构酶转录表达的原理示意图。
[0015]
图2:pgi基因敲除片段的构建及验证电泳图。其中,m:marker;1:pgi上游同源臂;2:pgi下游同源臂;3:重叠片段;4:阴性对照5:敲除后鉴定片段。
[0016]
图3:p
esar-p-esar整合片段构建及验证电泳图。其中:m:marker;1:上游同源臂;3:his g*基因片段;3:下游同源臂;4:重叠片段;5:原菌对照;6:阳性菌的鉴定片段。
[0017]
图4:p
esar-p-esai整合片段构建及验证电泳图。其中:m:marker;1:上游同源臂;3:p
esar-p-esai基因片段;3:下游同源臂;4:重叠片段;5:原菌对照;6:阳性菌的鉴定片段。
[0018]
图5:p
esas
整合片段构建及验证电泳图。其中:m:marker;1:上游同源臂;3:p
esas
基因片段;3:下游同源臂;4:重叠片段;5:原菌对照;6:阳性菌的鉴定片段。
[0019]
图6:p
trp
整合片段构建及验证电泳图。其中:m:marker;1:上游同源臂;3:p
trp
基因片段;3:下游同源臂;4:重叠片段;5:原菌对照;6:阳性菌的鉴定片段。
[0020]
图7:p
esar-p
整合片段构建及验证电泳图。其中:m:marker;1:上游同源臂;3:p
esar-p
基因片段;3:下游同源臂;4:重叠片段;5:原菌对照;6:阳性菌的鉴定片段。
[0021]
图8:p
trc-esar整合片段构建及验证电泳图。其中:m:marker;1:上游同源臂;3:p
trc-esar基因片段;3:下游同源臂;4:重叠片段;5:原菌对照;6:阳性菌的鉴定片段。
[0022]
图9:p
esar-p-esai整合片段构建及验证电泳图。其中:m:marker;1:上游同源臂;3:p
esar-p-esai基因片段;3:下游同源臂;4:重叠片段;5:原菌对照;6:阳性菌的鉴定片段。
具体实施方式
[0023]
下面通过具体的实施方案叙述本发明。除非特别说明,本发明中所用的技术手段均为本领域技术人员所公知的方法。另外,实施方案应理解为说明性的,而非限制本发明的范围,本发明的实质和范围仅由权利要求书所限定。对于本领域技术人员而言,在不背离本发明实质和范围的前提下,对这些实施方案中的物料成分和用量进行的各种改变或改动也属于本发明的保护范围。
[0024]
第一方面,本发明提供了一种大肠杆菌基因工程菌株,该基因工程菌株引入大肠杆菌色氨酸操纵子调控pgi基因的转录表达,包括:以大肠杆菌色氨酸操纵子的启动子p
trp
替换了pgi基因的原有启动子。因启动子p
trp
的强度受控于阻遏蛋白trpr和效应分子色氨酸的调控,进而使pgi基因的转录表达也受到trpr和色氨酸的调控。
[0025]
根据本发明,优选的,所述启动子p
trp
的核苷酸序列如seq id no:1所示。
[0026]
根据本发明,为了进一步提高组氨酸的生产效率,优选的,该基因工程菌株还引入以酰基高丝氨酸内酯(n-acyl-homoserine lactone,ahl)作为信号分子介导的群体感应(quorum sensing,qs)系统调控trpr基因的转录表达,包括:引入ahl合成酶基因esai和群体感应转录调控因子esar的编码基因esar,并且以启动子p
esar-p
替换了trpr基因原有启动子。
[0027]
本发明的调控原理如图1所示。总体上来说,在菌体生长期,trpr基因无法转录,
pgi基因可正常转录表达;当菌体生长到一定程度后,trpr基因开始转录表达,产物trpr与色氨酸的复合体可结合在启动子p
trp
的调控区域,终止pgi基因的转录表达。
[0028]
优选的,所述ahl合成酶基因esai的核苷酸序列如seq id no:2所示,其ncbi genbank:l32183.1。
[0029]
优选的,所述基因esar的核苷酸序列如seq id no:3所示,其ncbi genbank:l32184.1。
[0030]
优选的,所述ahl合成酶基因esai连接有启动子p
esar-p
,所述启动子p
esar-p
的核苷酸序列如seq id no:4所示。
[0031]
优选的,所述基因esar连接有启动子p
trc
,所述启动子p
trc
的核苷酸序列如seq id no:5所示。
[0032]
根据本发明,用于构建所述大肠杆菌基因工程菌株的出发菌株可以是任意的大肠杆菌,根据本发明一种优选的实施方式,所述出发菌株是一株工程菌e.coli why2-3(该工程菌及其构建过程已公布于发明专利cn111321102a),所述e.coli why2-3是以e.coli w3110为出发菌株过表达大肠杆菌组氨酸操纵子基因hisd、hisb、hisc、hish、hisa、hisf和hisi,以及异源过表达核苷酸序列如seq id no:6所示的谷氨酸棒杆菌atp转磷酸核糖基酶hisg突变体编码基因hisg*而获得。
[0033]
第二方面,本发明提供如上所述的大肠杆菌基因工程菌株的构建方法,该方法包括:在大肠杆菌中,以色氨酸操纵子的启动子p
trp
替换了pgi基因原有启动子;可选的引入ahl合成酶基因esai和群体感应转录调控因子esar的编码基因esar,并且以启动子p
esar-p
替换了trpr基因原有启动子。
[0034]
本发明第一方面已经详细介绍了上述各基因的选择、启动子的选择、出发菌株的选择等,此处不再赘述第一方面已经介绍过的内容。
[0035]
根据本发明一种具体的实施方式,该方法包括:
[0036]
(1)在大肠杆菌why2-3中,使用基因编辑方法将pgi基因原有启动子p
pgi
替换为色氨酸操纵子的启动子p
trp
,获得菌株his rp-3;
[0037]
(2)在菌株his rp-3中,使用基因编辑方法将trpr基因原有启动子p
trpr
替换为启动子p
esar-p
,获得菌株his rp-4a;
[0038]
(3)在菌株his rp-4a中,使用基因编辑方法将esar基因与启动子p
trc
的融合片段p
trc-esar整合在yjgx基因位点,获得菌株his rp-4b;
[0039]
(4)在菌株his rp-4b中,使用基因编辑方法将esai基因与启动子p
esar-p
的融合片段p
esar-p-esai整合在yciq基因位点,获得菌株his rp-4。
[0040]
进一步的,所述大肠杆菌why2-3是从e.coli w3110出发经过如下改造步骤后获得:在e.coli w3110中,使用基因编辑的方法,将基因hisg*与启动子p
trc
的融合片段p
trc-hisg*整合在tdcd基因位点和ylbe基因位点,以及将e.coli w3110组氨酸操纵子基因hisdbchafi(包含hisd、hisb、hisc、hish、hisa、hisf和hisi七个基因)与启动子p
trc
的融合片段p
trc-hisdbchafi整合在yghx基因位点。
[0041]
第三方面,本发明提供如上所述的大肠杆菌基因工程菌株在高产组氨酸中的用途,包括:在适宜条件培养所述大肠杆菌基因工程菌株,并从其培养物中收集组氨酸。
[0042]
根据本发明一种优选的实施方式,所述基因工程菌株his rp-4用于发酵生产组氨
酸的用途,经过摇瓶发酵24h后,组氨酸的产量为6.5g/l,糖酸转化率为11.7%,单位菌体产量0.339g/l,相比出发菌株why2-3分别提高了109.7%、200%和242.4%。
[0043]
根据本发明一种优选的实施方式,所述适宜条件是指培养温度37℃,200r/min振荡培养,维持ph在7.0-7.2,并且培养基组成为:葡萄糖20-40g/l,酵母提取物2-5g/l,蛋白胨5-7g/l,nano
3 15-17g/l,kh2po
4 1-3g/l,k2hpo
4 5-7g/l,mgso4·
7h2o 1-2g/l,柠檬酸钠1-2g/l,feso4·
7h2o 20-40mg/l,mnso4·
7h2o 5-15mg/l,v
b1
、v
b3
、v
b5
、v
b12
、vh各1-3mg/l,其余为水,ph 7.0-7.2。
[0044]
以下将通过具体的实施例对本发明进行更详细描述。以下实施例中:
[0045]
如无特别说明,本发明实施例中所涉及的基因编辑方法参照文献(li y,lin z,huang c,et al.metabolic engineering of escherichia coli using crispr
–
cas9 meditated genome editing.metabolic engineering,2015,31:13-21.)或发明专利cn111321102a进行,其他所涉及的分子生物学、基因工程等具体操作手段根据本领域人员容易获得的技术手册、教科书或文献报道均可以实现。
[0046]
实施例1:大肠杆菌基因工程菌株his rp-1的构建
[0047]
敲除组氨酸工程菌株why2-3的pgi基因,构建菌株his rp-1。
[0048]
(1)首先,出发菌株why2-3是从e.coli w3110出发经过如下改造步骤后获得:在e.coli w3110中,使用基因编辑的方法,将基因hisg*(seq id no:6)与启动子p
trc
的融合片段p
trc-hisg*整合在tdcd基因位点和ylbe基因位点,以及将e.coli w3110组氨酸操纵子基因hisdbchafi(包含hisd、hisb、hisc、hish、hisa、hisf和hisi七个基因)与启动子p
trc
的融合片段p
trc-hisdbchafi整合在yghx基因位点。具体的操作步骤可以参照cn111321102a的实施例1进行。
[0049]
(2)菌株构建过程所涉及的引物见下表:
[0050]
引物序列(5
’‑3’
)pgi-up-sacgctaacggcactaaaaccapgi-up-agctgaccgttagtgcctggtgcgttgacttccggcattapgi-dn-staatgccggaagtcaacgcaccaggcactaacggtcagcpgi-dn-atctttatcatctttcagctctggcgrna-pgi-sagtcctaggtataatactagtttctgacctcggcccatacagttttagagctagaagrna-pgi-attctagctctaaaactgtatgggccgaggtcagaaactagtattatacctaggact
[0051]
(3)菌株构建的具体过程
[0052]
以e.coli w3110基因组为模板,根据其pgi基因的上下游序列设计上游同源臂引物(pgi-up-s、pgi-up-a)和下游同源臂引物(pgi-dn-s、pgi-dn-a),并pcr扩增其上下游同源臂片段。上述片段通过重叠pcr的方法获得pgi基因的敲除片段(上游同源臂-下游同源臂),构建pgrb-pgi使用的含靶序列的dna片段通过引物grna-pgi-s和grna-pgi-a的退火制得。将该重组片段和质粒pgrb-pgi电转至why2-3的感受态细胞,筛选阳性菌株后再将质粒消除获得菌株his rp-1。pgi敲除片段的构建和阳性菌株的pcr验证的电泳图见附图2。其中,上游同源臂的长度应为450bp,下游同源臂的长度应为450bp,整合片段的总长应为900bp,pcr验证时,阳性菌pcr扩增片段长度应为900bp,原菌pcr扩增片段长度应为1670bp。
[0053]
实施例2:大肠杆菌基因工程菌株his rp-2的构建
[0054]
在出发菌株why2-3中引入pantoea stewartii subsp.stewartii的群体感应系统调控pgi基因的表达,构建菌株his rp-2。
[0055]
(1)将p
trc-esar片段整合在菌株why2-3的yjgx基因位点;
[0056]
以e.coli w3110基因组为模板,根据其yjgx基因的上下游序列设计上游同源臂引物(yjgx-up-s、yjgx-up-a)和下游同源臂引物(yjgx-dn-s、yjgx-dn-a),并pcr扩增其上下游同源臂片段;并根据esar基因(seq id no:3)设计引物(esar-s、esar-a),然后再扩增esar基因片段。启动子p
trc
(seq id no:5)则设计在上游同源臂的下游引物和esar基因的上游引物中。上述片段通过重叠pcr的方法获得esar基因的整合片段(上游同源臂-p
trc-esar-下游同源臂),构建pgrb-yjgx使用的含靶序列的dna片段通过引物yjgx-grna-s和yjgx-grna-a的退火制得。将该片段和质粒pgrb-trpr电转化至why2-3的感受态细胞,筛选阳性菌株后再将质粒消除获得菌株his rp-2a。p
trc-esar整合片段的构建和阳性菌株的pcr验证的电泳图见附图3。其中,上游同源臂的长度应为561bp,所扩增的esar基因片段长度应为873bp,下游同源臂的长度应为505bp,整合片段的总长应为1697bp,pcr验证时,阳性菌pcr扩增片段长度应为1697bp,原菌pcr扩增片段长度应为1286bp。
[0057]
(2)将p
esar-p-esai片段整合在菌株his rp-2a的yciq基因位点
[0058]
以e.coli w3110基因组为模板,根据其yciq基因的上下游序列设计上游同源臂引物(yciq-up-s、yciq-up-a)和下游同源臂引物(yciq-dn-s、yciq-dn-a),并pcr扩增其上下游同源臂片段;并根据esai基因(seq id no:2)设计引物(esai-s、esai-a),然后再扩增esai基因片段。启动子p
esar-p
(seq id no:4)则设计在上游同源臂的下游引物和esai基因的上游引物中。上述片段通过重叠pcr的方法获得esai基因的整合片段(上游同源臂-p
esar-p-esai-下游同源臂),构建pgrb-yciq使用的含靶序列的dna片段通过引物yciq-grna-s和yciq-grna-a的退火制得。将该片段和质粒pgrb-trpr电转化至his rp-2a的感受态细胞,筛选阳性菌株后再将质粒消除获得菌株his rp-2b。p
esar-p-esai整合片段的构建和阳性菌株的pcr验证的电泳图见附图4。其中,上游同源臂的长度应为646bp,所扩增的esai基因片段长度应为789bp,下游同源臂的长度应为640bp,整合片段的总长应为1895bp,设计鉴定引物并进行pcr验证,阳性重组子所扩增的片段长度应为1269bp,原菌无条带。
[0059]
(3)将pgi基因的启动子p
pgi
替换为p
esas
[0060]
以e.coli w3110基因组为模板,根据其pgi基因的上下游序列设计上游同源臂引物(pgi-up-s、pgi-up-a)和下游同源臂引物(pgi-dn-s、pgi-dn-a),并pcr扩增其上下游同源臂片段;并根据p
esas
基因(seq id no:7)设计引物(esas-s、esas-a),然后再扩增p
esas
基因片段。上述片段通过重叠pcr的方法获得p
esas
基因的整合片段(上游同源臂-p
esas-下游同源臂),构建pgrb-pgi使用的含靶序列的dna片段通过引物grna-pgi-s和grna-pgi-a的退火制得。将该片段和质粒pgrb-pgi电转化至his rp-2b的感受态细胞,筛选阳性菌株后再将质粒消除获得菌株his rp-2。p
esas
整合片段的构建和阳性菌株的pcr验证的电泳图见附图5。其中,上游同源臂的长度应为421bp,所扩增的p
esas
基因片段长度应为252bp,下游同源臂的长度应为531bp,整合片段的总长应为1115bp,pcr验证时,阳性菌pcr扩增片段长度应为1115bp,原菌pcr扩增片段长度应为1247bp。
[0061]
(4)上述菌株构建过程所涉及的引物见下表:
[0062]
[0063][0064]
实施例3:大肠杆菌基因工程菌株his rp-3的构建
[0065]
将组氨酸工程菌株why2-3中pgi基因的启动子p
pgi
替换为色氨酸启动子p
trp
,构建菌株his rp-3。
[0066]
(1)菌株构建的具体过程
[0067]
以e.coli w3110基因组为模板,根据其pgi基因的上下游序列设计上游同源臂引物(pgi-up-s、pgi-trp-up-a)和下游同源臂引物(pgi-trp-dn-s、pgi-dn-a),并pcr扩增其上下游同源臂片段;并根据trp操纵子基因设计引物(trp-s、trp-a),然后再扩增p
trp
基因片段(seq id no:1)。上述片段通过重叠pcr的方法获得p
trp
基因的整合片段(上游同源臂-p
trp-下游同源臂),构建pgrb-pgi使用的含靶序列的dna片段通过引物grna-pgi-s和grna-pgi-a的退火制得。将该片段和质粒pgrb-pgi电转化至why2-3的感受态细胞,筛选阳性菌株后再将质粒消除获得菌株his rp-3。p
trp
整合片段的构建和阳性菌株的pcr验证的电泳图见附图6。其中,上游同源臂的长度应为417bp,所扩增的p
trp
基因片段长度应为365bp,下游同
源臂的长度应为523bp,整合片段的总长应为1231bp,设计鉴定引物并进行pcr验证,阳性重组子所扩增的片段长度应为975bp,原菌无条带。
[0068]
(2)上述菌株构建过程所涉及的引物见下表:
[0069]
引物序列(5
’‑3’
)pgi-up-saaactggatgttgcggatagcpgi-trp-up-aacctgcacagccataccacaatcatcggctacaggggcttrp-sagcccctgtagccgatgattgtggtatggctgtgcaggttrp-agcgttggattgatgtttttcattgttattctctaattttgttcaaaapgi-trp-dn-sttttgaacaaaattagagaataacaatgaaaaacatcaatccaacgcpgi-dn-acagagcttcggtcaccatgtattrp-jd-scactcccgttctggataatgtttpgi-jd-agtttttcatcacctgccgctgrna-pgi-sagtcctaggtataatactagtctcaggtgttatcacaggacgttttagagctagaagrna-pgi-attctagctctaaaacgtcctgtgataacacctgagactagtattatacctaggact
[0070]
实施例4:大肠杆菌基因工程菌株his rp-4的构建
[0071]
在菌株his rp-3中引入pantoea stewartii subsp.stewartii的群体感应系统调控pgi基因的表达,构建菌株his rp-4。
[0072]
(1)将菌株his rp-3中trpr基因启动子p
trpr
替换为p
esar-p
[0073]
以e.coli w3110基因组为模板,根据其trpr基因的上下游序列设计上游同源臂引物(trpr-up-s、trpr-up-a)和下游同源臂引物(trpr-dn-s、trpr-dn-a),并pcr扩增其上下游同源臂片段。启动子p
esar-p
(seq id no:4)则设计在上游同源臂的下游引物和下游同源臂的上游引物中。上述片段通过重叠pcr的方法获得p
esar-p
基因的整合片段(上游同源臂-p
esar-p-下游同源臂),构建pgrb-trpr使用的含靶序列的dna片段通过引物grna-trpr-s和grna-trpr-a的退火制得。将该片段和质粒pgrb-trpr电转化至his rp-3的感受态细胞,筛选阳性菌株后再将质粒消除获得菌株his rp-4a。p
esar-p
整合片段的构建和阳性菌株的pcr验证的电泳图见附图7。其中,上游同源臂的长度应为552bp,下游同源臂的长度应为565bp,整合片段的总长应为1198bp,设计鉴定引物并进行pcr验证,阳性重组子所扩增的片段长度应为651bp,原菌无条带。
[0074]
(2)将p
trc-esar基因整合在菌株his rp-4a的yjgx基因位点
[0075]
以e.coli w3110基因组为模板,根据其yjgx基因的上下游序列设计上游同源臂引物(yjgx-up-s、yjgx-up-a)和下游同源臂引物(yjgx-dn-s、yjgx-dn-a),并pcr扩增其上下游同源臂片段;并根据esar基因(seq id no:3)设计引物(esar-s、esar-a),然后再扩增esar基因片段。启动子p
trc
则设计在上游同源臂的下游引物和esar基因的上游引物中。上述片段通过重叠pcr的方法获得esar基因的整合片段(上游同源臂-p
trc-esar-下游同源臂),构建pgrb-yjgx使用的含靶序列的dna片段通过引物yjgx-grna-s和yjgx-grna-a的退火制得。将该片段和质粒pgrb-trpr电转化至his rp-4a的感受态细胞,筛选阳性菌株后再将质粒消除获得菌株his rp-4b。p
trc-esar整合片段的构建和阳性菌株的pcr验证的电泳图见附图8。其中,上游同源臂的长度应为561bp,所扩增的esar基因片段长度应为873bp,下游同源臂的长度应为505bp,整合片段的总长应为1697bp,pcr验证时,阳性菌pcr扩增片段长度应
为1697bp,原菌pcr扩增片段长度应为1286bp。
[0076]
(3)将p
esar-p-esai基因片段整合在菌株his rp-4b的yciq基因位点
[0077]
以e.coli w3110基因组为模板,根据其yciq基因的上下游序列设计上游同源臂引物(yciq-up-s、yciq-up-a)和下游同源臂引物(yciq-dn-s、yciq-dn-a),并pcr扩增其上下游同源臂片段;并根据esai基因(seq id no:2)设计引物(esai-s、esai-a),然后再扩增esai基因片段。启动子p
esar-p
则设计在上游同源臂的下游引物和esai基因的上游引物中。上述片段通过重叠pcr的方法获得esai基因的整合片段(上游同源臂-p
esar-p-esai-下游同源臂),构建pgrb-yciq使用的含靶序列的dna片段通过引物yciq-grna-s和yciq-grna-a的退火制得。将该片段和质粒pgrb-trpr电转化至his rp-4b的感受态细胞,筛选阳性菌株后再将质粒消除获得菌株his rp-4。p
esar-p-esai整合片段的构建和阳性菌株的pcr验证的电泳图见附图9。其中,上游同源臂的长度应为646bp,所扩增的esai基因片段长度应为789bp,下游同源臂的长度应为640bp,整合片段的总长应为1895bp,设计鉴定引物并进行pcr验证,阳性重组子所扩增的片段长度应为1269bp,原菌无条带。
[0078]
(4)上述菌株构建过程所涉及的引物见下表:
[0079]
[0080][0081]
实施例5:菌株why2-3、his rp-1、his rp-2、his rp-3、his rp-4发酵生产组氨酸实验
[0082]
发酵生产组氨酸的方法:
[0083]
将菌种活化后制备种子液,按10-15%接种量接种到装有500ml三角瓶中(终体积为30ml),九层纱布封口,37℃,200r/min振荡培养,发酵过程中通过补加氨水维持ph在7.0-7.2;补加60%(m/v)葡萄糖溶液维持发酵进行。
[0084]
优选的发酵培养基组成为:葡萄糖20-40g/l,酵母提取物2-5g/l,蛋白胨5-7g/l,nano
3 15-17g/l,kh2po
4 1-3g/l,k2hpo
4 5-7g/l,mgso4·
7h2o 1-2g/l,柠檬酸钠1-2g/l,feso4·
7h2o 20-40mg/l,mnso4·
7h2o 5-15mg/l,v
b1
、v
b3
、v
b5
、v
b12
、vh各1-3mg/l,其余为水,ph 7.0-7.2。
[0085]
发酵结果分析:
[0086]
相较于出发菌株why2-3,基因工程菌株his rp-1、his rp-2、his rp-3、his rp-4分别采用了四种方法来调控pgi基因的活性。其中,菌株his rp-1中pgi基因缺失;菌株his rp-2中引入一种玉米病原菌pantoea stewartii subsp.stewartii的群体感应系统,使得pgi基因在生长前期表达,后期不表达;菌株his rp-3中pgi基因的启动子替换为p
trp
,使得pgi基因的转录表达受控于胞内色氨酸的水平;菌株his rp-4是在his rp-3基础上引入pantoea stewartii subsp.stewartii的群体感应系统,从而抑制调控蛋白trpr在生长前期的表达量,而在生长后期正常表达,利用色氨酸启动子和群体感应相结合的方式调控pgi基因的转录表达。对以上四株菌进行摇瓶培养24h后产组氨酸的相关效果如下表所示:
[0087][0088]
his rp-1相较出发菌why2-3 od
600
和组氨酸产量分别降低60.9%和51.6%,但是单位菌体产量提升24.2%,并且转化率也提高了48.7%。说明pgi基因的敲除明显抑制了菌株的生长,虽然产量受到明显影响,但是单位菌体产量和转化率的提升说明pgi基因的敲除
大幅降低了emp的通量,可促进菌体组氨酸合成途径的加强,结果也同之前的文献报道相符。
[0089]
his rp-2相较出发菌why2-3 od
600
基本保持不变,组氨酸产量和单位菌体产量分别降低48.3%和45.5%。通过对his rp-2和why2-3中pgi基因进行转录量的分析发现,虽然在发酵稳定期(24h),his rp-2的pgi基因的转录量较why2-3明显降低,但是在组氨酸生产关键时期(8-16h),his rp-2的pgi基因的转录量远远高于why2-3,说明直接利用所选择的群体感应系统直接调控pgi基因,不仅未能适时有效降低pgi基因的转录水平,反而在前期大幅提升了pgi基因的转录水平,和我们的预期目标相悖。
[0090]
his rp-3相较出发菌why2-3 od
600
降低56.7%,组氨酸产量和单位菌体产量分别提升32.2%和207.1%,转化率提升了82.1%。说明色氨酸启动子有效减弱了pgi基因的表达,虽然菌株生长受到明显抑制,但是组氨酸产量和生产强度有了大幅提升。
[0091]
his rp-4相较出发菌why2-3 od
600
降低38.4%,组氨酸产量、糖酸转化率和单位菌体产量分别提升109.7%、200%和242.4%;而相较his rp-3,his rp-4的od
600
提升42.2%,产量、糖酸转化率和单位菌体产量分别提升58.5%、64.8%和11.5%。说明本发明通过将色氨酸调控机制与群体感应系统相结合,实现了对pgi基因的自调控,在满足菌体基本生长的前提下,有效降低了pgi基因的转录表达,从而提升了组氨酸的产量、转化率和生产强度。
[0092]
虽然本发明已经以较佳实施例公开如上,但其并非用以限定本发明,任何本领域技术人员,在不脱离本发明的精神和原理的情况下,可以对这些实施例进行各种形式和细节的变化、修改、替换和变型,本发明的范围由权利要求及其等同物所限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1