一种FFPE样本RNA文库及其构建方法与流程
一种ffpe样本rna文库及其构建方法
技术领域
1.本发明涉及生物技术领域,尤其涉及一种ffpe样本rna文库及其构建方法。
背景技术:
2.离体的肿瘤组织容易丧失原有正常的结构和功能,而使用福尔马林固定石蜡包埋处理的样本(formalin-fixed andparrffin-embedded,ffpe)可以在常温保留很久,所以对肿瘤组织进行石蜡包埋保存就成了医院里最常用的样本储存手段。福尔马林的固定容易使组织中的核酸发生不同程度的降解和分子间的交联,石蜡的高温渗入过程进一步加速核酸的降解,保存的时间及环境对样品中的核酸也有巨大的影响,从石蜡切片中提取高质量核酸,并构建出信息完整、可用于后续rna研究的文库,是相当的难题。
3.rnaffpe样本的复杂性:1.会与蛋白质和其他细胞成分交联;2.随着时间的推移可导致福尔马林氧化成甲酸,从而引起碱基脱嘌呤和核酸链断裂,造成提取量和rna质量偏低;3.易受到pcr抑制剂的污染,样本中含有pcr抑制剂。其复杂性导致了基于扩增子方法学建库测序的rna文库构建困难,出库量较低,因而建库成功率低;引物和接头二聚体较严重,因而文库质量差。
4.illuminaminiseq和miseq测序平台对上机文库变性有最低要求为2nm,低于2nm变性效果差,测序成功率低;因此规定了可用于上机文库测序的文库需要具有一定的出库浓度(例如本专利所涉及的panel要求rna文库出库浓度≥0.4ng/μl);另外,由于rna样本的复杂性,低质量的ffpe rna样本建库过程中容易引入过高的引物二聚体和接头二聚体。因此,在临床应用中,常规扩增子建库方法文库质量低,建库成功率低。为了解决扩增子文库质量(提高文库目的片段比例,降低引物和接头二聚体等非主峰片段比例)和rna建库成功率低(rna出库浓度较低)的问题,科技工作者提出了诸如增加扩增循环数,提高测序深度等方法。
5.现有技术的缺陷和不足:
6.1、增加扩增循环数,可提高rna建库成功率,但低质量的ffpe rna样本建库过程中容易引入过高的引物二聚体(dimer)和接头二聚体,过高的接头二聚体会造成采用qubit定量的方法文库主峰定量的不准确,影响文库的准确pooling。
7.2、增加测序深度,直接提高了单样本检测成本,造成不必要的浪费,并且质量差的ffpe样本由于出库浓度低并不能进行illumina平台的测序。
技术实现要素:
8.有鉴于此,本发明提供了一种ffpe样本rna文库的构建方法。该方法在建库过程中,加入一段人造序列(seq id no:1所示的dna片段)与混有质粒上下游引物的可用于检测rna fusion突变的引物池panel一起进行有效扩增和建库,提高了建库成功率和rna扩增子文库质量。
9.为了实现上述发明目的,本发明提供以下技术方案:
10.本发明提供一种ffpe样本rna文库的构建方法,包括如下步骤:
11.步骤1:提取ffpe样本的rna,经反转录获得cdna产物;
12.步骤2:向所述cdna产物的扩增体系中加入含seq id no:1所示dna片段的质粒和用于扩增seq id no:1所示dna片段的引物;按照rna测序文库的构建方法构建文库。
13.本发明在建库过程中掺入一段dna片段人造序列,即包含seq id no:1所示片段的质粒,与混有seq id no:2~3上下游引物的可用于检测rna fusion突变的引物池panel一起进行有效扩增和建库。该dna片段与人类基因组没有同源序列,可以是人工合成,也可以以spikein-09质粒为模板由seq id no:2~3所示的引物扩增获得。各序列如下:
14.seq id no.1:
15.tgtgaactgtcatcggtccgatcaattagtctagtgtgcgttattcagatcgagtgagtacatgattcgtcagtgtggatcaattacagttaggccgctgacacattagtaacgtcggcaagcacttagtcgtgtcgtaagccagtgtgtcgtgtct。
16.上游引物1:
[0017]5’‑
cctacacgacgctcttccgatcttgtgaactgtcatcggtccg-3’[0018]
(seq id no.2);
[0019]
下游引物2:
[0020]5’‑
ttcagacgtgtgctcttccgaaagacacgacacactggctt-3’(seq id no.3)。
[0021]
其中,所述上下游引物中5’端下划线标记的序列为tail序列,这部分序列可以与本发明第二轮pcr中的index引物序列结合,形成完整的接头,用于完成测序。
[0022]
一些实施方案中,dna产物的扩增体系,掺入的spikein-09质粒的拷贝数为4.5
×
10^5。
[0023]
步骤1中,在所述反转录之前还包括对rna进行预处理的步骤;所述预处理为:将所述rna与反转录混合液混合,65℃反应5min,获得预处理反应产物。
[0024]
一些实施方案中,步骤2中,所述rna测序文库的构建方法包括:对所述加入seq id no:1所示dna片段的扩增体系进行第一轮pcr扩增,经纯化、消化后,回收消化产物进行第二轮pcr扩增,纯化,获得ffpe样本rna文库。
[0025]
一些实施方案中,步骤2中,所述第一轮pcr扩增的扩增体系包括:
[0026]
1μl spikein质粒、4μl扩增混合液1,5nm~10nm检测rna fusion突变的引物池,5nm~10nm seq id no:2~3所示的引物,10μl~30cdna产物,无核酸酶水补足至20μl。
[0027]
一些实施方案中,所述扩增混合液1包括dna聚合酶、缓冲液、镁离子和dntp。
[0028]
一些实施方案中,所述上、下游引物的比例为1:1~2:1。一些具体实施例中,上、下游引物的比例为1:1。
[0029]
以上扩增体系中,各组分的浓度均为工作浓度。
[0030]
第一轮pcr扩增的程序为:预变性95℃10min,变性98℃15s,退火/延伸60℃5min,保存10℃∞;变性、退火/延伸为10个循环。
[0031]
本发明步骤2中,采用磁珠对第一轮pcr扩增产物和第二pcr扩增产物进行纯化,然后用te buffer洗脱。
[0032]
一些实施方案中,所述消化体系包括:
[0033]
消化缓冲液2μl,消化反应液1μl,第一轮pcr扩增后的纯化产物10μl,无核酸酶水7
μl。
[0034]
本发明中,消化缓冲液和消化反应液为本领域常见的种类,具体实施例中采用的消化缓冲液和消化反应液为paragon genomics专利产品。
[0035]
一些实施方案中,所述第二轮pcr扩增的扩增体系包括:
[0036]
8μl扩增混合液2,20.4μl无核酸酶水,10μl消化纯化后反应产物,400~1000nm index引物混合液;
[0037]
所述index引物混合液包括index上游引物和index下游引物,index上下游引物比例为1:1。所述引物为seq id no:4~5,各引物序列如下:
[0038]
index上游引物:
[0039]
caagcagaagacggcatacgagatcttccttcgtgactggagttcagacgtgtgctcttccgatc*t(seq id no.4)
[0040]
注:下划线标记的碱基为barcode序列,可替换;*表示3’末端的碱基t进行磷酸化修饰。
[0041]
index下游引物:
[0042]
aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatc*t(seq id no.5)。
[0043]
注:*表示3’末端的碱基t进行磷酸化修饰。
[0044]
扩增混合液2包括dna聚合酶、缓冲液、镁离子和dntp。
[0045]
第二轮pcr扩增的程序为:预变性95℃10min,变性98℃15s,60℃退火/延伸75s,保存10℃∞;变性、退火/延伸为10个循环。
[0046]
扩增混合液1和2为pcr扩增试剂,包括dna聚合酶、缓冲液、镁离子和dntp。本发明对扩增混合液的具体来源没有限制,具体实施例中,采用的扩增混合液1和2为paragon genomics专利产品。
[0047]
本发明第二轮pcr扩增的扩增体系中,所述index引物混合物浓度为400~1000nm,具体可为400nm、700nm或1000nm。
[0048]
以上扩增体系中各组分的浓度均为工作浓度。
[0049]
本发明中,所述第二轮pcr扩增采用的index引物与第一轮pcr中的引物池panel和seq id no:2~3所示的引物形成完整的接头引物。
[0050]
本发明还提供了由本发明以上构建方法获得的rna文库。
[0051]
与常规方法相比,本发明具有如下优点:
[0052]
1、本发明能够降低rna扩增子文库引物二聚体以及非特异扩增产物的形成,使文库能更准确的定量;
[0053]
2、能够提升rna扩增子文库出库量,能够满足低质量ffpe样本达到illumina测序平台最低变性浓度的要求,使临床中低质量的样本能够进入到测序和检测分析;适于推广应用。
附图说明
[0054]
图1示本发明方法与常规方法文库2100数据比较。
具体实施方式
[0055]
本发明提供了一种ffpe样本rna文库及其构建方法。本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
[0056]
本发明采用的试材皆为普通市售品,皆可于市场购得。
[0057]
下面结合实施例,进一步阐述本发明:
[0058]
实施例1
[0059]
(1)3例ffpe rna的提取以及定量,样本编号分别为(r001,r002,r003),提取试剂盒选用无锡臻悦生物科技有限公司生产的ffpe dna/rna共提取试剂盒;
[0060]
(2)spikein质粒的配制:质粒用ph 8.0te buffer稀释至4.5
×
10^5copies/μl备用;
[0061]
(3)引物的配制:spikein质粒上下游引物按一定比例稀释成终浓度为50nm的spikein质粒引物母液备用,比例为1:1;
[0062]
(4)rna样本预处理:在200μl pcr管配制反应体系:反转录混合液5μl,每个rna样本50ng,用灭菌去离子水补齐到10μl,将上述反应管于65℃反应5min。
[0063]
(5)逆转录反应及纯化:在200μl pcr管配制反应体系:2μl反转录酶混合液,8μl反转录缓冲液2,10μl预处理反应产物;将上述反应管放入pcr仪器,运行反转录程序:25℃10min,42℃15min,70℃15min。纯化采用beckmanxp磁珠2.2
×
,纯化后用10μl te buffer洗脱备用。
[0064]
(6)mpcr反应及纯化:在200μl pcr管配反应体系:1μl spikein质粒溶液,4μl扩增混合液1,5nm引物池panel,5nm spikein质粒引物母液,10μl cdna产物,3μl无核酸酶水;将上述反应管放入pcr仪器,运行mpcr程序:预变性95℃10min,变性98℃15s,退火/延伸60℃5min,保存10℃∞;变性,退火/延伸为10个循环。纯化采用1.3
×
beckman xp磁珠,纯化后用10μl tebuffer洗脱备用。
[0065]
(7)消化反应及纯化:在200μl pcr管配反应体系:2μl消化缓冲液,1μl消化反应液;10μl mpcr后纯化产物,7μl无核酸酶水;将上述反应管放入pcr仪器,37℃精确孵育10min。纯化采用1.3
×
beckman xp磁珠,纯化后用10μl tebuffer洗脱备用。
[0066]
(8)2
nd
pcr反应及纯化:在200μl pcr管配反应体系:8μl扩增混合液2,20.4μl无核酸酶水,10μl消化纯化后反应产物,400nm index引物混合液;将上述反应管放入pcr仪器,运行pcr程序预变性95℃10min,变性98℃15s,退火/延伸75s,保存10℃∞;变性,退火/延伸为10个循环。纯化采用1
×
beckmanxp磁珠,纯化用20μlte buffer洗脱备用。
[0067]
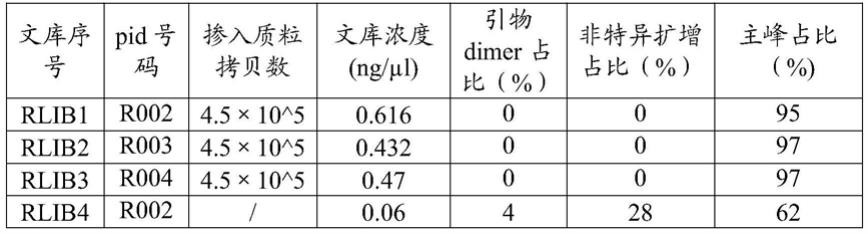
(9)文库定量及质检:用qubit仪器对文库进行浓度检测;用agilent tapestation 2100对文库进行文库片段大小检测;本发明方法(文库编号rlib1~rlib3)与常规方法(注:常规方法为未加spikein质粒与spikein质粒引物,其余与本发明方法一致,文库编号rlib4~rlib6)建库的文库质控数据比较见表1,本发明方法与常规方法2100文库峰图见图1。
[0068]
表1本发明方法与常规方法文库质控数据比较
[0069][0070][0071]
以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1