一种高产多杀菌素J/L的工程菌及其构建方法与应用与流程
一种高产多杀菌素j/l的工程菌及其构建方法与应用
技术领域
1.本发明涉及一种高产多杀菌素j/l的工程菌及其构建方法与应用,属于生物技术领域。
背景技术:
2.多杀菌素是从刺糖多孢菌(saccharopolyspora spinosa)发酵液中提取得到的一种大环内酯类生物杀虫剂,具有无公害、高效、低毒、易降解等特点。它对非靶标动物无害,对哺乳动物也无致癌作用。多杀菌素是一种混合物,在该混合物中,多杀菌素a和d是主要组分并且对关键昆虫目标具有最高活性。多杀菌素j和l(多杀菌素混合物中的两种次要组分)是乙基多杀菌素(第二代多杀菌素杀虫剂)的前体。乙基多杀菌素是5,6-二氢-3
’‑
乙氧基多杀菌素j(主要组分)和3
’‑
乙氧基多杀菌素l的混合物(参见说明书附图图1),其是从多杀菌素j/l进行乙基化和氢化两步化学合成得到的,因此,能够高产多杀菌素j/l的发酵菌株是工业化生产乙基多杀菌素的关键。从多杀菌素生物合成途径分析,将spnk基因失活后,代谢流会发生改变,多杀菌素的发酵菌株将积累更多的多杀菌素j/l,而不是多杀菌素a/d,从而就可以得到大量的合成乙基多杀菌素的前体。
3.中国专利文献cn103119152a公开了修饰spnk基因以消除其表达的3
’‑
o-甲基转移酶活性的方法,利用基因工程同源重组或是通过诱变或利用rna干扰技术(rnai)对spnk基因进行修饰使其失活,从而得到多杀菌素j/l的生产菌株。然而,这几种方法均存在各种缺陷:通过同源重组使spnk失活,需要两步筛选,效率非常低;通过诱变筛选得到spnk的基因突变株,需要更大规模的筛选,而且专一性差;rnai无法完全抑制基因的转录或翻译,并且容易脱靶导致假阳性或假阴性结果。
4.crispr/cas是新一代的基因组定点编辑技术,能对活细胞dna进行精准操作,实现基因片段的特异性插入,删除,替换等,利用crispr/cas技术可以改变基因的序列和功能,控制细胞的命运和生命特征,为遗传性疾病的治疗提供新的方法。crispr/cas系统存在于几乎所有的古菌和大多数细菌中。crispr/cas由一系列cas蛋白(cas1、cas2、cas4和效应蛋白如cas9、cpf1等)的编码基因和一段crispr序列组成,后者由一段前导序列、许多重复序列和间隔序列顺序排列组成。根据cas基因的组成和效应蛋白的数量,crispr被分为了2类5型,共16种亚型。1类为利用多个效应蛋白复合物干扰靶基因的crispr/cas系统,包括ⅰ、ⅲ和ⅳ型;2类为利用单一的效应蛋白干扰靶基因的crispr/cas系统,包括ⅱ型和
ⅴ
型。目前研究得最为清楚的crispr/cas9为2类ⅱ型crispr系统,2015年张锋小组新发现的crispr/cpf1属于2类
ⅴ
型crispr系统。其中ⅱ类系统是目前使用最多且最简单的rna指导核酸内切酶技术,该系统主要包含两个成分:sgrna和cas9,其中sgrna(single-guide rna)是由细菌的内源性crispr rna(crrna)和反式作用crispr rna(trans-activating crispr rna,tracrrna)融合而成的人工合成rna,主要发挥引导作用;cas9由hnh和ruvc两个核酸酶结构域组成,可完成对外源dna序列的识别和切割。
5.近年来,cpf1酶的基因编辑技术迅速发展,由于其具有更高水平的靶标特异性,
cpf1酶成为了应用于较多宿主的基因编辑工具。cpf1是一种v型的ii类crispr内切核酸酶,其除了具有dna切割活性外,还具有rna酶活性,可以将crispr rna(crrna)共转录物加工成独立的成熟crrna。此外,与cas9不同的是,cpf1识别位于靶序列5’端富含胸腺嘧啶(t)的pam(原间隔序列临近基序,protospacer-adjacent motif,简称pam)序列,而cas9则识别位于靶序列的3’端的富含鸟嘌呤(g)的pam序列(5
’‑
ngg-3’)。cpf1识别的pam序列与cas9不同,极大地扩宽了crispr系统基因组编辑的靶点范围,尤其是富含at的基因组。crispr/cpf1的开发有利于突破和克服crispr/cas9应用中的一些限制,因此被称为是新一代的crispr基因组编辑工具。失去dnase酶活性的cpf1(dnase-dead cpf1,ddcpf1),仍保留了加工pre-crrna的功能。将作为基因工程表达调控元件的ddcpf1引入合成生物学研究领域可以更精准地靶向目的基因,也具有更强大的调控功能。与crispr/dcas9类似,在应用crispr/ddcpf1时,crrna的靶向位置与其调控基因表达的能力有着明显关联,同样也可以通过选择不同的靶位点来控制基因调控的强度。
6.crispr/ddcpf1系统中的ddcpf1蛋白分子量相对较大,对宿主细胞的代谢是一种较大的负担,因此,它们能否在特定宿主细胞中进行表达是难以预期的。也有另外的可能性是,其虽然可以在宿主细胞中表达但所表达的ddcpf1蛋白没有功能。因此,crispr/ddcpf1系统能否应用于特定的宿主环境是需要进行试验和探索的,目前,现有技术中还没有检索该技术的应用。
技术实现要素:
7.针对现有技术的不足,本发明提供了一种高产多杀菌素j/l的工程菌及其构建方法与应用。
8.本发明的技术方案如下:
9.一种高产多杀菌素j/l的工程菌,所述工程菌是以刺糖多孢菌ql-1为出发菌株,通过插入crrna抑制刺糖多孢菌ql-1的spnk基因表达至失活,构建得到。
10.根据本发明优选的,所述spnk基因如seq id no.1所示,所述crrna的序列如seq id no.2、seq id no.3或seq id no.4所示。
11.上述高产多杀菌素j/l的工程菌的构建方法,是以spnk基因作为靶基因,采用crispr/ddcpf1技术设计抑制刺糖多孢菌ql-1的spnk基因表达的crrna,然后将crrna连接至表达质粒上,再转化大肠杆菌得到大肠杆菌转化子,最后将大肠杆菌转化子接合转移至刺糖多孢菌中,检测发酵产物后获得。
12.根据本发明优选的,所述高产多杀菌素j/l的工程菌的构建方法,具体步骤如下:
13.(1)以spnk基因作为靶基因,在开放阅读框内搜索cpf1的原间隔序列临近基序识别靶基因序列的tttn或ttn,选取原间隔序列临近基序之后的23个核苷酸为原间隔序列,即得到抑制刺糖多孢菌ql-1的spnk基因表达的crrna,具体核苷酸序列如seq id no.2、seq id no.3或seq id no.4所示;
14.(2)采用直接合成法或引物退火法将步骤(1)所述crrna连接到含有ddcpf1编码基因的质粒载体pql-2上,得到重组质粒pql-spnk-crrna;
15.(3)将构建好的重组质粒pql-spnk-crrna转化大肠杆菌感受态细胞,得到大肠杆菌转化子菌液;
16.(4)将大肠杆菌转化子菌液与刺糖多孢菌的菌丝悬液混合后涂r6平板培养基,培养6~10天后选择接合子进行培养,经筛选后获得高产多杀菌素j/l的基因工程菌,记为ql-1/pql-spnk-crrna。
17.根据本发明优选的,步骤(2)中,所述直接合成法的具体步骤如下:按照crrna的序列采用同源重组方法合成含有crrna的插入序列(含有spei酶切位点),再将含有crrna的插入序列连接至质粒载体pql-2的spei酶切位点处,得到重组质粒pql-spnk-crrna。
18.根据本发明优选的,步骤(2)中,所述引物退火法的具体步骤如下:
19.以crrna为模板进行pcr扩增,得到crrna序列,所述pcr扩增的引物序列如下:
20.crrna-f:
[0021]5′‑
atttctactgttgtagatnnnnnnnnnnnnnnnnnnnnnnnactagtgcgtcgatatct-3
′
;
[0022]
crrna-r:
[0023]5′‑
agatatcgacgcactagtnnnnnnnnnnnnnnnnnnnnnnnatctacaacagtagaaat-3
′
;
[0024]
其中“n”代表如seq id no.2、seq id no.3或seq id no.4所示的crrna;
[0025]
pcr扩增程序:变性,95℃2min;95℃开始每50秒下降0.5℃,降至25℃(140个循环),最后4℃保温;
[0026]
pcr体系:anneaking buffer for dna oligos(5
×
)20μl,crrna-f(50μm)20μl,crrna-r(50μm)20μl,ddh2o 40μl,总体积100μl。
[0027]
将质粒载体pql-2和pcr用spei酶单酶切后,经组装酶连接至质粒载体pql-2的spei酶切位点处,得到重组质粒pql-spnk-crrna。
[0028]
根据本发明优选的,步骤(4)中,所述大肠杆菌转化子菌液与刺糖多孢菌的菌丝悬液的体积比为(1~10):(1~10)。
[0029]
根据本发明优选的,步骤(4)中,所述r6平板培养基成分为(g/l):蔗糖200.0,糊精10.0,酪蛋白氨基酸1.0,mgso4·
7h2o 0.05,*谷氨酸钠盐11.0,k2so
4 0.1,*cacl2·
2h2o 7.0,*mops(0.1mol/l,ph7.2)100.0,*微量元素(ml)1.0ml,琼脂(sigma agar)20.0;
[0030]
微量元素组成为(mg/l):zncl
2 40,fecl3·
6h2o 200,cucl2·
2h2o 10,mncl2·
4h2o 10,na2b4o4·
10h2o 10,(nh4)6mo7o
24
·
4h2o 10。注:标示*的部分均分开灭菌,灭菌后再合并。
[0031]
上述基因工程菌在生产多杀菌素j/l中的应用。
[0032]
根据本发明优选的,所述应用,包括步骤如下:
[0033]
将基因工程菌ql-1/pql-spnk-crrna接种于的斜面培养基中,25~30℃下培养6~10天,收集菌丝,接种于种子培养基中,在25~30℃下培养2~4天,最后将种子液按照8~12%的体积比接种至发酵培养基中,在25~30℃,230~250r/min下培养2~4天,得发酵液。
[0034]
根据本发明优选的,所述斜面培养基组分如下:葡萄糖0.3%,蛋白胨0.5%,牛肉膏0.3%,氯化钠0.5%,琼脂2.7%,ph 7.0,均为质量百分比。
[0035]
根据本发明优选的,所述种子培养基组分如下:葡萄糖3.0%,可溶性淀粉1.0%,棉籽饼粉2.0%,黄豆饼粉0.2%,酵母粉0.2%,玉米浆1.0%,碳酸钙0.5%,ph 7.0,均为质量百分比。
[0036]
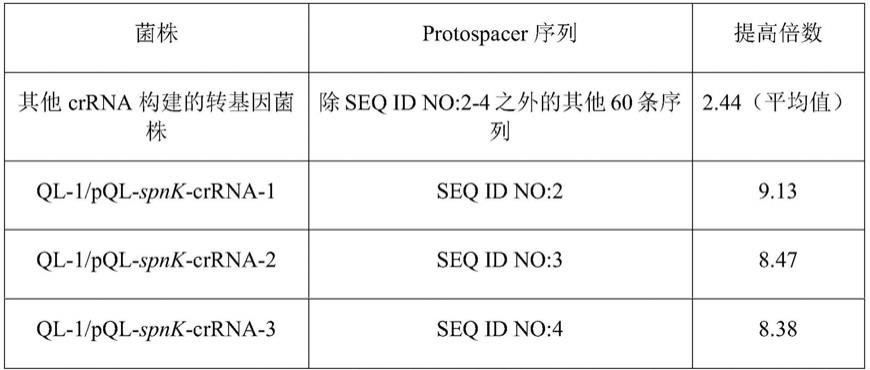
根据本发明优选的,所述发酵培养基组分如下:葡萄糖6.0%,可溶性淀粉3.0%,葵花油0.5%,棉籽饼粉2.0%,鱼粉蛋白胨1.0%,酵母粉0.2%,玉米浆1.0%,碳酸钙
0.5%,ph 7.0,均为质量百分比。
[0037]
本发明的技术特点及有益效果:
[0038]
1、本发明spnk基因作为靶基因,采用crispr/ddcpf1技术设计了3个抑制刺糖多孢菌ql-1的spnk基因表达的crrna,然后以刺糖多孢菌ql-1作为出发菌株,通过插入crrna抑制刺糖多孢菌ql-1的spnk基因表达至失活,构建得到高产多杀菌素j/l的ql-1/pql-spnk-crrna基因工程菌。本发明首次在刺糖多孢菌中引入crispri技术,通过crispr/ddcpf1方法对spnk基因进行了抑制失活,从而实现了多杀菌素j/l的高水平表达,进而达到了高产多杀菌素j/l的目的。
[0039]
2、本发明构建的ql-1/pql-spnk-crrna工程菌可以稳定传代,并且能够显著提高刺糖多孢菌多杀菌素j/l的产量,是野生刺糖多孢菌的8~9倍。
附图说明
[0040]
图1为乙基多杀菌素的化学结构。
[0041]
图2为spnk的原间隔序列设计简图。
[0042]
图3为引物退火法构建pql-spnk-sgrna质粒。
[0043]
图4为野生刺糖多孢菌ql-1多杀菌素a/d的hplc分析图谱。
[0044]
图5为基因工程菌ql-1/pql-spnk-crrna-1产多杀菌素j/l的hplc分析图谱。
[0045]
图6为基因工程菌ql-1/pql-spnk-crrna-2产多杀菌素j/l的hplc分析图谱。
[0046]
图7为基因工程菌ql-1/pql-spnk-crrna-3产多杀菌素j/l的hplc分析图谱。
[0047]
图8为基因工程菌ql-1/pql-spnk-crrna-4产多杀菌素j/l的hplc分析图谱。
[0048]
图9为基因工程菌ql-1/pql-spnk-crrna-5产多杀菌素j/l的hplc分析图谱。
具体实施方式
[0049]
下面结合实施例和附图对本发明的技术方案作进一步说明,但是本发明的保护范围并不仅限于此。实施例中涉及的试剂及药品,若无特殊说明,均为普通市售产品;实施例中涉及的实验操作,若无特殊说明,均为本领域常规操作。
[0050]
实施例1重组crispr/ddcpf1-crrna质粒的构建
[0051]
如图2所示,将spnk基因作为靶基因(序列如seq id no.1所示),在spnk基因的orf内搜索cpf1酶的pam识别序列tttn或ttn,选取pam序列之后的23个核苷酸视为原间隔序列(protospacer)。通过手动设计评估protospacer的脱靶风险,将spnk基因orf中的所有protospacer列出,共获得63条protospacer序列用于筛选评估,此63条protospacer序列分别命名为crrna-1至crrna-63,经后续验证得知,crrna-1、crrna-2和crrna-3对spnk基因的抑制效果最好,其序列分别如seq id no.2、seq id no.3和seq id no.4所示;crrna-4、crrna-5作为后续实施例的的对照组,其序列分别如seq id no.5和seq id no.6所示,其余crrna本发明不做进一步说明。
[0052]
以crrna-1为例,采用直接合成法或引物退火法将crrna-1连接到含有ddcpf1编码基因的质粒载体pql-2上,得到重组质粒pql-spnk-crrna-1。
[0053]
所述直接合成法的具体步骤如下:按照crrna的序列采用同源重组方法合成含有crrna的插入序列(含有spei酶切位点),具体合成由。。。。公司完成,再将含有crrna的插入
序列连接至质粒载体pql-2的spei酶切位点处,得到重组质粒pql-spnk-crrna-1。
[0054]
含有crrna的插入序列的核苷酸序列如下:
[0055]5′‑
tgcccacaacagcatcgcggtgccacgtgtggaccgcgtcggtcagatcctccccgcacctctcgccagccgtcaagatcgaccgcgtgcacctgcgatcgccgatcaaccgcgactagcatcgggcgcaagccgccactcgaacggacactcgcatgcatactagagggatcctgttcacattcgaaccgtctctgctttgacaacatgctgtgcggtgttgtaaagtcgtggccaaatttctactgttgtagatggtggtcaggtcggccaggctcgactagtgcgtcgatatctcgtaggtacccttgattaattaagcatagtttaaactcaccaataaaaaacgcccggcggcaaccgagcgttctgaacaaatccagatggagttctgaggtcattactggatgtacacccgaattcgtaatcatgtcatagctgtttcctgtgtgaaattgttatccgctcacaattccacacaacatacgagccggaagcataaagtgtaaagcctggggtgcctaatgagtgagctaactcacattaattgcgttgcgctcactgcccgctttccagtcgggaaacctgtcgtgccagct-3
′
。
[0056]
如图3所示,所述引物退火法的具体步骤如下:以crrna为模板进行pcr扩增,得到crrna序列,所述pcr扩增的引物序列如下:
[0057]
crrna-1-f:
[0058]5′‑
atttctactgttgtagatggtggtcaggtcggccaggctcgactagtgcgtcgatatct-3
′
;
[0059]
crrna-1-r:
[0060]5′‑
agatatcgacgcactagtggtggtcaggtcggccaggctcgatctacaacagtagaaat-3
′
;
[0061]
pcr扩增程序:变性,95℃2min;95℃开始每50秒下降0.5℃,降至25℃(140个循环),最后4℃保温;
[0062]
pcr体系:anneaking buffer for dna oligos(5
×
)20μl,crrna-1-f(50μm)20μl,crrna-1-r(50μm)20μl,ddh2o 40μl,总体积100μl。
[0063]
将质粒载体pql-2和pcr用spei酶单酶切后,经组装酶连接至质粒载体pql-2的spei酶切位点处,得到重组质粒pql-spnk-crrna-1。
[0064]
按照相同的方法制备得到重组质粒pql-spnk-crrna-2至重组质粒pql-spnk-crrna-63。
[0065]
实施例2转化子及基因工程菌的构建
[0066]
以重组质粒pql-spnk-crrna-1进行高产多杀菌素j/l的基因工程菌的构建,具体方法如下:
[0067]
(1)大肠杆菌的转化
[0068]
将实施例1制备的重组质粒pql-spnk-crrna-1转化感受态大肠杆菌s17-1(购自takara公司),然后挑取转化子至4ml lb培养基(apr 100μg/ml)的小试管中37℃振荡培养12小时后,再按2%接种量转接于50ml lb的250ml三角瓶,37℃振荡培养2小时左右,使菌液od值在0.4~0.6之间,将菌液移入50ml的无菌塑料离心管,离心(4000rpm,10min,4℃),倒去上清,菌体用20ml lb洗涤2次(4000rpm,10min,4℃),最后重悬于1~2ml lb中,得到大肠杆菌转化子菌液;
[0069]
(2)转化子与刺糖多胞菌ql-1接合转移
[0070]
将刺糖多孢菌ql-1划线接种于斜面培养基中,在30℃下培养7天,然后从斜面挑取适量菌体于50ml tsb培养基中培养72小时左右达到对数生长期,再以1%接种量转接于50ml tsb培养45小时,使菌液达到对数生长后期,离心倒去上清得到菌丝体。菌丝体用20ml
的lb液体洗涤2次(4000rpm,10min,4℃),最后重悬于20ml lb,得到刺糖多孢菌的菌丝悬液。将大肠杆菌菌液与菌丝悬液按体积比(10∶1、1∶1、1∶10)于ep管中混匀。将混合菌液涂r6平板培养基,用涂棒充分混匀菌液,28℃恒温箱培养,培养20小时后,取出平板培养基涂抗生素(940μl ddh2o+100μl amp+50μl萘啶酸)平板,再于30℃恒温箱继续培养一周后,得到接合子;
[0071]
所述r6平板培养基成分为(g/l):蔗糖200.0,糊精10.0,酪蛋白氨基酸1.0,mgso4·
7h2o 0.05,*谷氨酸钠盐11.0,k2so
4 0.1,*cacl2·
2h2o 7.0,*mops(0.1mol/l,ph7.2)100.0,*微量元素(ml)1.0ml,琼脂(sigma agar)20.0。注:标示*的部分均分开灭菌,灭菌后再合并。
[0072]
微量元素组成为(mg/l):zncl
2 40,fecl3·
6h2o 200,cucl2·
2h2o 10,mncl2·
4h2o 10,na2b4o4·
10h2o 10,(nh4)6mo7o
24
·
4h2o 10。
[0073]
所述斜面培养基组分如下:葡萄糖0.3%,蛋白胨0.5%,牛肉膏0.3%,氯化钠0.5%,琼脂2.7%,ph 7.0,均为质量百分比。
[0074]
(3)工程菌的筛选
[0075]
挑取接合子于含有阿伯拉霉素(50μg/ml)的tsb中培养,然后将菌液涂于含有阿伯拉霉素(50μg/ml)的斜面培养基,28℃培养,获得目标基因工程菌ql-1/pql-spnk-crrna-1。
[0076]
按照相同的方法制备得到基因工程菌ql-1/pql-spnk-crrna-2至基因工程菌ql-1/pql-spnk-crrna-63。
[0077]
实施例3采用基因工程菌ql-1/pql-spnk-crrna制备多杀菌素j/l
[0078]
以基因工程菌ql-1/pql-spnk-crrna-1为例进行多杀菌素j/l的制备,具体步骤如下:
[0079]
挑取实施例2中的基因工程菌ql-1/pql-spnk-crrna-1接种于相同的斜面培养基中,在28℃下培养8天后,接种于相同种子培养液中,在28℃下培养3天后,转接于发酵培养基中,在28℃,240r/min培养9天后放瓶,得发酵液。
[0080]
所述种子培养基组分如下:葡萄糖3.0%,可溶性淀粉1.0%,棉籽饼粉2.0%,黄豆饼粉0.2%,酵母粉0.2%,玉米浆1.0%,碳酸钙0.5%,ph 7.0,均为质量百分比。
[0081]
所述发酵培养基组分如下:葡萄糖6.0%,可溶性淀粉3.0%,葵花油0.5%,棉籽饼粉2.0%,鱼粉蛋白胨1.0%,酵母粉0.2%,玉米浆1.0%,碳酸钙0.5%,ph 7.0,均为质量百分比。
[0082]
按照相同的方法分别对野生刺糖多孢菌ql-1、基因工程菌ql-1/pql-spnk-crrna-2至基因工程菌ql-1/pql-spnk-crrna-63进行发酵培养,分别得到发酵液。
[0083]
将以上发酵液分别用甲醇超声,离心后取上清液做hplc分析。其中,hplc的流动相:0.2%乙酸铵∶甲醇=10∶90;流速:1ml/min;柱温:40℃;检测波长:254nm;进样量:5μl;分析柱:agilent c18。多杀菌素a和d出峰时间分别为约7.07min,8.399min,多杀菌素j和l出峰时间分别为约5.063min,5.883min。
[0084]
野生刺糖多孢菌ql-1多杀菌素a/d hplc分析图谱如图4所示,基因工程菌ql-1/pql-spnk-crrna-1产多杀菌素j/l hplc分析图谱如图5所示,基因工程菌ql-1/pql-spnk-crrna-2产多杀菌素j/l hplc分析图谱如图6所示,基因工程菌ql-1/pql-spnk-crrna-3产多杀菌素j/l hplc分析图谱如图7所示,基因工程菌ql-1/pql-spnk-crrna-4产多杀菌素j/
l hplc分析图谱如图8所示,基因工程菌ql-1/pql-spnk-crrna-5产多杀菌素j/l hplc分析图谱如图9所示。
[0085]
由此可见,可见通过crispr/ddcpf1系统能够实现在刺糖多孢菌中进行基因干扰,该系统可以在刺糖多孢菌中实现基因表达水平调控的操作,并且通过失活spnk基因成功地抑制了多杀菌素a/d的表达并提高了多杀菌素j/l的产量;而ql-1/pql-spnk-crrna-1,ql-1/pql-spnk-crrna-2和ql-1/pql-spnk-crrna-3三株菌株则具有出人意料的多杀菌素j/l高表达量,具体数据比较结果参见表4。
[0086]
表4基因工程菌相比于野生型菌株的多杀菌素j/l表达量提高倍数
[0087][0088]
由表4可知,pql-spnk-crrna质粒的转入可有效实现刺糖多孢菌ql-1的多杀菌素a/d表达的抑制,多杀菌素j/l表达量的提升,进而达到了高产多杀菌素j/l的目的。在由63条crrna构建的所有基因工程菌ql-1/pql-spnk-crrna中,有三个菌株产多杀菌素j/l效果最好,分别是基因工程菌ql-1/pql-spnk-crrna-1、基因工程菌ql-1/pql-spnk-crrna-2和基因工程菌ql-1/pql-spnk-crrna-3。而基因工程菌ql-1/pql-spnk-crrna-4和基因工程菌ql-1/pql-spnk-crrna-5的多杀菌素j/l的产量要明显低于基因工程菌ql-1/pql-spnk-crrna-1、基因工程菌ql-1/pql-spnk-crrna-2和基因工程菌ql-1/pql-spnk-crrna-3,其余的58种基因工程菌更低,本发明不做进一步说明。野生刺糖多孢菌ql-1的多杀菌素j/l的产量最低,主要产出多杀菌素a/d。
[0089]
实施例4基因工程菌株的传代稳定性
[0090]
分别挑取实施例2和3中能够高产多杀菌素j/l的菌株,经过十次传代,再在实施例2和3中相同的斜面培养基中培养,经过与实施例2和3相同的种子培养基及发酵培养基,发酵结果显示本发明所构建的多杀菌素基因工程菌具有良好的传代稳定性,十次传代后的菌株依然保持较高的发酵水平。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1