一种手性二胺配位金属催化剂的制备方法与流程
1.本发明涉及催化剂领域,尤其涉及一种手性二胺配位金属催化剂。
背景技术:
2.手性二胺类化合物是一类重要的合成中间体,其在医学、农用化学以及工业生产中都有着广泛的应用。现阶段,手性二胺类化合物在不对称催化领域取得了较大的突破,由手性环己二胺、1,2-二苯基乙二胺等简单的手性二胺类化合物,衍生出了诸多从简单到复杂的二胺类化合物;同时手性二胺类化合物作为手性配体,将其与金属配位得到的手性二胺配位金属催化剂,被广泛地应用于酮与亚胺的不对称催化氧化、不对称diels-alder反应等。相比于小分子手性二胺催化剂,手性二胺配位金属催化剂具有更多的活性位点,具有反应速度快、高收率、高选择性等优势,使其具有更高的实用价值。
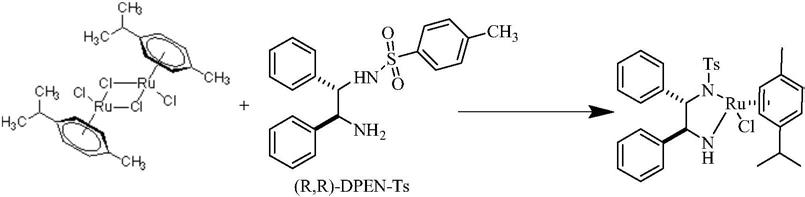
3.目前,{[(1r,2r)-(-)-2-氨基-1,2-二苯基乙基](对甲苯磺酰基)氨基)}(对异丙基甲苯)氯化钌的合成工艺是以配体和金属化合物直接配位反应,这样的反应制备过程极为繁琐,且所得粗产品会包含脱氯手性物质,得到的产品纯度较低,因此需进一步重结晶提纯,导致效率降低,产品收率降低,金属耗量增大。
技术实现要素:
[0004]
为了解决上述技术问题,本发明提供了一种手性二胺配位金属催化剂的制备方法。本发明以对伞花烃氯化钌二聚体作为金属前驱体,于混合溶剂体系下,通过碱试剂的配合,直接与配体加热反应制备得到{[(1r,2r)-(-)-2-氨基-1,2-二苯基乙基](对甲苯磺酰基)氨基)}(对异丙基甲苯)氯化钌,与现有技术相比,不仅反应条件温和,产物以晶体形式析出,无需进一步重结晶提纯,缩短了制备流程,大大降低了生产成本,且所得产物收率高(>95.0%)、纯度高(>98.5%)。
[0005]
本发明的具体技术方案为:一种手性二胺配位金属催化剂的制备方法,包括以下步骤:(a)于室温条件下,将配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺和有机溶剂a混合,搅拌至澄清,得到溶液体系a;所述有机溶剂a为二氯甲烷、甲醇、乙醇和异丙醇中一种或两种。
[0006]
(b)于30~60℃条件下,将对伞花烃氯化钌二聚体和有机溶剂b混合,搅拌均匀;再加入碱试剂,搅拌均匀,得到溶液体系b;所述有机溶剂b为石油醚、环己烷和甲苯中一种或两种;所述有机溶剂b的体积为有机溶剂a的1.0~3.0倍。所述碱试剂为三乙胺、氢氧化钠和氢氧化钾中的一种,进一步优选为氢氧化钠。
[0007]
(c)在搅拌条件下,将步骤(a)所得溶液体系a滴加到步骤(b)所得溶液体系b中,30~75℃下搅拌反应3.0~8.0h,反应结束后,分离得到固体,真空干燥,得到目标产物{[(1r,2r)-(-)-2-氨基-1,2-二苯基乙基](对甲苯磺酰基)氨基)}(对异丙基甲苯)氯化钌。
[0008]
本发明制备方法的化学反应过程示意图如下:
如背景技术部分所述,现有技术是将配体和金属化合物直接配位反应,该方法反应制备过程极为繁琐,得到的产品纯度较低,且须进一步重结晶提纯,导致产品收率降低,金属耗量增大。为此,本发明以对伞花烃氯化钌二聚体作为金属前驱体,于混合溶剂体系下,通过碱试剂的配合,直接与配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺加热反应制备得到{[(1r,2r)-(-)-2-氨基-1,2-二苯基乙基](对甲苯磺酰基)氨基)}(对异丙基甲苯)氯化钌,与现有技术相比,反应条件温和,产物以晶体形式析出,无需进一步重结晶提纯,不仅缩短了制备流程,大大降低了生产成本,且所得产物收率高(>95.0%)、纯度高(>98.5%),金属利用率得到提升。
[0009]
要实现上述技术效果,本发明制备方法的关键点主要在于以下几个方面:(1)本发明采用混合溶剂体系,即在步骤(a)和(b)中分别选用了溶剂a和溶剂b,并且严格限定了溶剂a和溶剂b的种类以及相对用量。这是由于本发明团队在研究过程中发现,选择混合溶剂比单一溶剂有更好的选择性和更高的产率。究其原因,溶剂效应对反应速度和产物组成影响很大,当溶剂分子结构与反应机理相适应时,就会有较高的反应活性和选择性。在本发明中,极性的溶剂a有更好的溶解度,且有利于配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺脱氢,形成阴离子(结构如下图),在碱试剂的作用下,促使其与原料对伞花烃氯化钌二聚体进行反应;非极性的溶剂b的加入,可改变反应环境,有利于合成目标产物。综上,本发明通过混合溶剂,使反应体系极性适中,由于产物是芳香烃物质,调节非极性惰性溶剂比率,可进一步提高反应收率。
[0010]
(2)本发明在步骤(b)中采用特定几个种类的碱试剂。由于本发明的反应体系会产生酸性物质,通过添加碱试剂,可调节反应体系ph值,促进原料对伞花烃氯化钌二聚体脱氯,加快其与配体的反应。进一步地,本发明团队在试验中偶然发现,与其他碱试剂相比,选用本发明上述特定几类碱试剂的效果更佳。我们在发现此差异后分析原因,可能是:对于氢氧化钾等无机碱,对于碱中的阳离子,特别是碱金属在反应中的作用,这些金属离子可能与反应中间体有相互作用,或是其本身就是反应活性物种的一部分,因此它们对反应进程有着重要影响.金属离子的半径大小、电子云密度、与活性催化物种的相互作用方式都可能是影响反应的原因;对于三乙胺等有机碱,相对无机碱,其碱性相对较弱,但其溶解度更有优势,能有效消除反应过程所产生的酸,促进反应的进行。
[0011]
作为优选,所述有机溶剂a为乙醇;所述有机溶剂b为石油醚。乙醇为极性溶剂,且比其他有机溶剂更低的去质子电位,在碱试剂的配合下,有利于目标产物的生成;石油醚为
非极性溶剂,改变反应体系环境,进一步提高目标产物的收率。
[0012]
作为优选,步骤(a)中,所述配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺与有机溶剂a的用量比为1g∶(4.0~10.0)ml。
[0013]
有机溶剂a选择合适的用量,有利于配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺的溶解,投入量过少,导致配体溶解不完全,不利于反应体系均一化;投入量过多,溶剂极性较大,可溶解部分目标产物,影响反应收率。本发明团队通过研究发现上述用量较为合适。
[0014]
作为优选,步骤(b)中,所述对伞花烃氯化钌二聚体与有机溶剂b的用量比为1g∶(5.0~30.0)ml。
[0015]
有机溶剂b为非极性溶剂,选择合适比例的非极性惰性溶剂的用量,调节反应溶液的极性条件,有利于提高目标产物的收率。作为优选,步骤(b)中,所述碱试剂的摩尔质量为对伞花烃氯化钌二聚体的3.0~6.0倍。
[0016]
作为优选,所述配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺的摩尔质量为对伞花烃氯化钌二聚体的2.05~2.6倍。
[0017]
选择合适的配体用量,促使原料对伞花烃氯化钌二聚体反应完全,但配体用量过多,会使部分配体被产物包裹,影响目标产物的纯度。
[0018]
作为优选,步骤(a)-(c)在无氧条件下进行。
[0019]
为了进一步提高产物收率和纯度,本发明所有步骤均在无氧条件下进行。
[0020]
作为优选,步骤(b)中,将对伞花烃氯化钌二聚体和有机溶剂b混合后,搅拌5~15min;加入碱试剂后,搅拌10~30min。
[0021]
作为优选,步骤(c)中,所述溶液体系a的滴加时间为15~60min。
[0022]
选择有效合理的时间,在碱试剂的作用下,调节反应体系环境,促使反应完全。
[0023]
作为优选,步骤(c)中,所述真空干燥的温度40-80℃,时间为3.0-10.0h,真空度状态至<-0.05mpa。
[0024]
与现有技术相比,本发明具有以下技术效果:(1)本发明将金属前驱体,于混合溶剂体系下,通过碱试剂的配合,直接与配体加热反应制备得到手性二胺配位金属催化剂,反应条件温和,产物以晶体形式析出,无需进一步重结晶提纯,制备流程短,大大降低了生产成本。
[0025]
(2)本发明方法制得的产物收率高(>95.0%)、纯度高(>98.5%)。
[0026]
(3)本发明整个反应过程不涉及危险化学品,操作环境更好更安全,适合工业化生产。
具体实施方式
[0027]
下面结合实施例对本发明作进一步的描述。
[0028]
总实施例一种手性二胺配位金属催化剂的制备方法,包括以下步骤:(a)于室温条件下、无氧环境中,按用量比1g∶(4.0~10.0)ml将配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺和有机溶剂a(选自二氯甲烷、甲醇、乙醇和异丙醇,最优选为乙醇)混合,搅拌至澄清,得到溶液体系a。
[0029]
(b)于30~60℃条件下、无氧环境中,按用量比1g∶(5.0~30.0)ml将对伞花烃氯化钌二聚体(配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺的摩尔质量为对伞花烃氯化钌二聚体的2.05~2.6倍)和有机溶剂b(选自石油醚、环己烷和甲苯,最优选为石油醚,体积为有机溶剂a的1.0~3.0倍)混合,搅拌5~15min;再加入摩尔质量为对伞花烃氯化钌二聚体的3.0~6.0倍的碱试剂(选自三乙胺、氢氧化钠和氢氧化钾,最优选为氢氧化钠),搅拌10~30min,得到溶液体系b。
[0030]
(c)在搅拌、无氧条件下,将步骤(a)所得溶液体系a滴加以到步骤(b)所得溶液体系b中,滴加时间为15~60min,30~75℃下搅拌反应3.0~8.0h,反应结束后,分离得到固体,真空干燥(40-80℃,3.0-10.0h,<-0.05mpa),得到目标产物{[(1r,2r)-(-)-2-氨基-1,2-二苯基乙基](对甲苯磺酰基)氨基)}(对异丙基甲苯)氯化钌。
[0031]
实施例1(a)于20~25℃室温条件下,无氧氛围中,将15.7g配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺和65ml无水乙醇混合,搅拌至澄清,得到溶液体系a;(b)于30℃条件下,无氧氛围中,将12.25g对伞花烃氯化钌二聚体和70ml石油醚混合,搅拌10min,再加入2.5g氢氧化钠,搅拌20min,得到溶液体系b;(c)在搅拌、无氧状态下,将步骤(a)得到的溶液体系a滴加到步骤(b)得到的溶液体系b中,滴加15min,60℃搅拌反应4.0h,反应结束后,冷却、过滤、洗涤、抽干,真空干燥(60℃,4.0h,-0.05mpa)得到目标产物24.41g,收率95.9%,产物纯度98.9%。
[0032]
本实施例制备的化合物的元素分析结果为c58.46%,h5.67%,ru15.72%;理论值为c58.52%,h5.55%,ru15.89%。
[0033]
实施例2(a)于20~25℃室温条件下,无氧氛围中,将16.7g配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺和90ml二氯甲烷混合,搅拌至澄清,得到溶液体系a;(b)于40℃条件下,无氧氛围中,将12.25g对伞花烃氯化钌二聚体和120ml环己烷混合,搅拌5min,再加入4.5g氢氧化钾,搅拌25min,得到溶液体系b;(c)在搅拌、无氧状态下,将步骤(a)得到的溶液体系a滴加到步骤(b)得到的溶液体系b中,滴加30min,40℃搅拌反应7.0h,反应结束后,冷却、过滤、洗涤、抽干,真空干燥(65℃,3.5h,-0.07mpa)得到目标产物24.23g,收率95.2%,产物纯度98.6%。
[0034]
本实施例制备的化合物的元素分析结果为c58.42%,h5.73%,ru15.67%;理论值为c58.52%,h5.55%,ru15.89%。
[0035]
实施例3(a)于20~25℃室温条件下,无氧氛围中,将17.5g配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺和120ml异丙醇混合,搅拌至澄清,得到溶液体系a;(b)于60℃条件下,无氧氛围中,将12.25g对伞花烃氯化钌二聚体和200ml甲苯混合,搅拌15min,再加入12.0g三乙胺,搅拌10min,得到溶液体系b;(c)在搅拌、无氧状态下,将步骤(a)得到的溶液体系a滴加到步骤(b)得到的溶液体系b中,滴加50min,75℃搅拌反应3.5h,反应结束后,冷却、过滤、洗涤、抽干,真空干燥(75℃,3.0h,-0.06mpa)得到目标产物24.31g,收率95.5%,产物纯度98.7%。
[0036]
本实施例制备的化合物的元素分析结果为c58.45%,h5.71%,ru15.68%;理论值
为c58.52%,h5.55%,ru15.89%。
[0037]
实施例4(a)于20~25℃室温条件下,无氧氛围中,将18.4g配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺和160ml甲醇混合,搅拌至澄清,得到溶液体系a;(b)于45℃条件下,无氧氛围中,将12.25g对伞花烃氯化钌二聚体和360ml石油醚混合,搅拌10min,再加入4.5g氢氧化钠,搅拌15min,得到溶液体系b;(c)在搅拌、无氧状态下,将步骤(a)得到的溶液体系a滴加到步骤(b)得到的溶液体系b中,滴加60min,65℃搅拌反应4.0h,反应结束后,冷却、过滤、洗涤、抽干,真空干燥(50℃,7.0h,-0.08mpa)得到目标产物24.36g,收率95.7%,产物纯度98.7%。
[0038]
本实施例制备的化合物的元素分析结果为c58.46%,h5.70%,ru15.68%;理论值为c58.52%,h5.55%,ru15.89%。
[0039]
实施例5(a)于20~25℃室温条件下,无氧氛围中,将16.0g配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺和40ml无水乙醇/40ml二氯甲烷混合,搅拌至澄清,得到溶液体系a;(b)于35℃条件下,无氧氛围中,将12.25g对伞花烃氯化钌二聚体和150ml石油醚混合,搅拌15min,再加入3.0g氢氧化钠,搅拌15min,得到溶液体系b;(c)在搅拌、无氧状态下,将步骤(a)得到的溶液体系a滴加到步骤(b)得到的溶液体系b中,滴加20min,45℃搅拌反应6.0h,反应结束后,冷却、过滤、洗涤、抽干,真空干燥(65℃,4.0h,-0.06mpa)得到目标产物24.28g,收率95.4%,产物纯度98.6%。
[0040]
本实施例制备的化合物的元素分析结果为c58.43%,h5.74%,ru15.67%;理论值为c58.52%,h5.55%,ru15.89%。
[0041]
实施例6(a)于20~25℃室温条件下,无氧氛围中,将17.0g配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺和80ml异丙醇/40ml二氯甲烷混合,搅拌至澄清,得到溶液体系a;(b)于40℃条件下,无氧氛围中,将12.25g对伞花烃氯化钌二聚体和150ml石油醚/100ml环己烷混合,搅拌10min,再加入6.0g氢氧化钾,搅拌20min,得到溶液体系b;(c)在搅拌、无氧状态下,将步骤(a)得到的溶液体系a滴加到步骤(b)得到的溶液体系b中,滴加35min,40℃搅拌反应7.5h,反应结束后,冷却、过滤、洗涤、抽干,真空干燥(65℃,4.0h,-0.07mpa)得到目标产物24.26g,收率95.3%,产物纯度98.6%。
[0042]
本实施例制备的化合物的元素分析结果为c58.42%,h5.69%,ru15.67%;理论值为c58.52%,h5.55%,ru15.89%。
[0043]
实施例7(a)于20~25℃室温条件下,无氧氛围中,将18.0g配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺和100ml乙醇/60ml异丙醇混合,搅拌至澄清,得到溶液体系a;(b)于50℃条件下,无氧氛围中,将12.25g对伞花烃氯化钌二聚体和160ml环己烷/200ml甲苯混合,搅拌10min,再加入10.0g三乙胺,搅拌10min,得到溶液体系b;(c)在搅拌、无氧状态下,将步骤(a)得到的溶液体系a滴加到步骤(b)得到的溶液体系b中,滴加45min,70℃搅拌反应3.5h,反应结束后,冷却、过滤、洗涤、抽干,真空干燥(75
℃,3.5h,-0.07mpa)得到目标产物24.31g,收率95.5%,产物纯度98.6%。
[0044]
本实施例制备的化合物的元素分析结果为c58.45%,h5.70%,ru15.67%;理论值为c58.52%,h5.55%,ru15.89%。
[0045]
实施例8(a)于20~25℃室温条件下,无氧氛围中,将650.0g配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺和3.0l无水乙醇混合,搅拌至澄清,得到溶液体系a;(b)于50℃条件下,无氧氛围中,将490.0g对伞花烃氯化钌二聚体和6.0l石油醚混合,搅拌10min,再加入128.0g氢氧化钠,搅拌15min,得到溶液体系b;(c)在搅拌、无氧状态下,将步骤(a)得到的溶液体系a滴加到步骤(b)得到的溶液体系b中,滴加20min,70℃搅拌反应3.5h,反应结束后,冷却、过滤、洗涤、抽干,真空干燥(70℃,3.0h,-0.06mpa)得到目标产物981.47g,收率96.4%,产物纯度98.9%。
[0046]
本实施例制备的化合物的元素分析结果为c58.45%,h5.69%,ru15.72%;理论值为c58.52%,h5.55%,ru15.89%。
[0047]
将各实施例的检测数据进行对比,结果如下表所示:案例收率纯度实施例195.9%98.9%实施例295.2%98.6%实施例395.5%98.7%实施例495.7%98.7%实施例595.4%98.6%实施例695.3%98.6%实施例795.5%98.6%实施例896.4%98.9%对比例1为了说明本发明中反应过程中无氧条件的重要性,在本案例中,反应过程处于有氧氛围下进行反应。取实施例1,无氧条件更改为通入空气,其他步骤不变,真空干燥得到目标产物22.37g,收率87.9%,产物纯度97.4%。说明反应过程处于有氧条件下,可能使配体变质或反应过程有副产物的生成,并且对目标产物的收率及纯度都有较大影响。
[0048]
对比例2为了说明本发明中反应过程中混合溶剂的重要性,在本案例中,反应过程中步骤(a)与步骤(b)使用相同溶剂。取实施例1,步骤(a)与步骤(b)都使用无水乙醇,其他步骤不变,反应所得物质须进一步重结晶提纯,真空干燥得到目标产物14.38g,收率只有56.5%,产物纯度98.6%(重结晶纯度)。说明反应步骤(a)与步骤(b)采用同一溶剂时,可能使配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺与对伞花烃氯化钌二聚体发生副反应,合成不含氯手性配体,须进一步提纯,分离副产物。
[0049]
对比例3为了说明本发明中反应过程中混合溶剂中溶剂用量的重要性,在本案例中,反应过程中步骤(a)增加溶剂a的用量。取实施例1,步骤(a)所用无水乙醇增加至180ml,其他步骤不变,反应所得物质须进一步重结晶提纯,真空干燥得到目标产物16.65g,收率只有
65.4%,产物纯度98.7%(重结晶纯度)。说明反应步骤(a)增加无水乙醇的用量,使配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺与对伞花烃氯化钌二聚体发生副反应,合成不含氯手性配体,须进一步提纯,分离副产物。
[0050]
对比例4为了说明本发明中反应过程中混合溶剂中溶剂用量的重要性,在本案例中,反应过程中步骤(b)增加溶剂b的用量。取实施例1,步骤(b)所用石油醚增加至380ml,其他步骤不变,反应所得物质须进一步重结晶提纯,真空干燥得到目标产物17.92g,收率只有70.4%,产物纯度98.7%(重结晶纯度)。说明反应步骤(b)增加石油醚的用量,导致反应体系极性下降,使配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺与对伞花烃氯化钌二聚体反应不完全,须进一步提纯,分离杂质。
[0051]
对比例5为了说明本发明中反应过程中碱试剂的重要性,在本案例中,反应过程处于无碱试剂条件下进行反应。取实施例1,不加碱试剂,其他步骤不变,过滤所得物质,进一步重结晶后,真空干燥得到目标产物13.67g,收率只有53.7%,产物纯度98.8%(重结晶纯度)。说明反应处于无碱试剂条件下,可能是反应难以进行或反应极为缓慢。
[0052]
对比例6为了说明本发明中反应过程中碱试剂的重要性,在本案例中,反应过程更换其他碱试剂进行反应。取实施例1,加碱试剂碳酸钠6.5g,其他步骤不变,过滤所得物质,真空干燥得到目标产物20.59g,收率只有80.9%,产物纯度95.6%。说明反应处于其他碱试剂条件下,反应可以进行,但收率有明显下降。
[0053]
对比例7为了说明本发明反应过程的优越性,在本案例中,采用金属化合物与配体直接反应的工艺方法。具体过程如下:第一步:于40℃条件下,将5.23g水合三氯化钌和100ml无水乙醇混合,搅拌10min,再加入8.18g水芹烯,升温至75℃搅拌反应4h,得到反应体系a;第二步:将配体(1r,2r)-n-(2-氨基-1,2-二苯乙基)对甲苯磺酰胺7.8g和50ml无水乙醇混合,搅拌15min,再加入到反应体系a中,继续搅拌反应8h;第三步:反应结束后,冷却、过滤、洗涤、抽干,所得粗产品进一步重结晶,真空干燥(65℃,4.0h,-0.06mpa)得到目标产物6.40g,收率50.3%,产物纯度98.9%(重结晶纯度)。
[0054]
将各实施例和对比例的反应条件和检测数据进行对比,结果如下表所示:案例与实施例1差异点收率纯度实施例1-95.9%98.9%对比例1有氧条件87.9%97.4%对比例2单一溶剂:无水乙醇56.5%98.6%(重结晶)对比例3溶剂a:无水乙醇增加至180ml65.4%98.7%(重结晶)对比例4溶剂b:石油醚增加至380ml70.4%98.7%(重结晶)对比例5不加碱试剂53.7%98.8%(重结晶)对比例6碱试剂:碳酸钠80.9%95.6%对比例7金属化合物与配体直接反应50.3%98.9%(重结晶)
由上表数据对比可知:在收率方面:对比例1-6在实施例1的基础上仅改变反应环境条件、溶剂、碱试剂,均会影响最终产物的收率。而对比例7是采用现有技术中常规的金属化合物与配体直接反应方法,收率也在较低水平。
[0055]
在纯度方面:对比例1是在有氧条件下进行,因此产物纯度降低,仅为97.4%;而对比例6改变碱试剂种类为碳酸钠,我们发现产物纯度急剧降低,仅为95.6%,可能原因是以碳酸钠作为碱试剂时导致产物中混有其他副产物;对比例2-5和7的纯度虽然较高,但是该纯度是经过重结晶后的结果,增加了反应步骤,而在未重结晶的情况下显然无法达到上述纯度水平。
[0056]
本发明中所用原料、设备,若无特别说明,均为本领域的常用原料、设备;本发明中所用方法,若无特别说明,均为本领域的常规方法。
[0057]
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制,凡是根据本发明技术实质对以上实施例所作的任何简单修改、变更以及等效变换,均仍属于本发明技术方案的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1