用于胶粘剂体系的官能化(共)聚合物的制作方法
用于胶粘剂体系的官能化(共)聚合物
1.本技术是基于申请日为2017年4月6日,申请号为201780034325.3,发明名称为:“用于胶粘剂体系的官能化(共)聚合物”的专利申请的分案申请。
技术领域
2.本发明涉及环氧官能化聚(甲基)丙烯酸酯,其特别地可用于作为胶粘剂组合物使用或作为胶粘剂组合物的组分。
背景技术:
3.目前,胶带以多种形式用作工艺中的辅助物和用于多种物体的粘合。在此,包括压敏胶粘剂组合物的自胶粘带具有永久粘性。它们可在无进一步固化的情况下典型地在粘合之后立即完成其粘合任务。这种自胶粘带有时可实现非常高的粘合强度。然而,在特定的应用中需要允许甚至更高粘合强度的胶粘剂解决方案。许多这种胶粘剂体系在热压制步骤中进行施加。在该情况下,它们熔融,润湿粘合基底并在冷却期间通过固化而形成强度。这样的胶粘剂体系另外还可具有化学反应性。以胶带形式使用的一些可热压制的胶粘剂体系在热压条件下显示不希望的挤出(“渗出”),因为熔融粘度随温度下降或者胶粘剂体系由于温度相关的内聚性损失而变得自由流动。在液体胶粘剂体系的情况下,挤出的问题已经在室温下发生。相反,在粘合过程中,需要良好的对待粘合基底的适应特性以实现胶粘剂层的最佳接触表面。这需要胶粘剂体系在压制条件下的一定程度的流动特性。
4.在本发明的上下文中,术语“挤出”或“渗出”(测试a)理解为是指未固化或不充分固化的胶粘剂组合物从胶粘剂层或胶粘粘合侧向逸出。这可以冷流的形式和/或在压力下和/或在升高的温度下发生并且是不合乎期望的。
5.此外,在反应性胶带的情况下,存在如下的要求:可在用于引发固化反应的活化时间方面快速加工胶粘剂体系。除了活化之外,固化也应快速进行到所需的固化程度。在本上下文中,特别有利的反应性体系是基于环氧化物的。
6.以胶带形式使用的可固化的基于环氧化物的胶粘剂体系常常含有成膜剂组分(其可为热塑性聚合物、弹性体或热塑性弹性体)以及通常由反应性树脂(在这些情况下是基于环氧化物的)和固化剂体系(也称为活化剂体系或引发剂体系)组成的反应性组分。实例将在ep 1 028 151 b1(聚(甲基)丙烯酸酯/低分子量环氧树脂)、ep 620 259 a1(聚酯/低分子量环氧树脂)、ep 721 975(聚烯烃/低分子量环氧树脂)和de 195 19 499 a1(热塑性聚氨酯/低分子量环氧树脂)中找到。
7.在本发明的上下文中,术语“可固化聚合物”、“可固化胶粘剂组合物(物质)”、或“可固化胶粘剂体系”理解为是指如下的聚合物或配制物:其包含可通过与作为额外刺激的升高的温度和/或辐射(特别地uv辐射)组合的固化剂组分的作用而参与反应的官能团,所述反应导致分子量的增加和/或聚合物或至少一种配制物成分的交联。
8.术语“固化剂”、“引发剂”、“活化剂”在本发明的上下文中同义地使用。它们描述如下的物质或物质混合物:其可在与升高的温度和/或辐射(特别地uv辐射)组合的情况下引
起涉及环氧官能团的固化反应。
9.此外,在将以反应性胶带形式使用的可固化胶粘剂体系的情况下,典型地必需确保在储存条件下足够的储存稳定性,使得反应性胶带可以简单的方式运输和储存,然后实际的反应性则仅在活化步骤中和在活化条件下发生/将要发生。在没有这种延迟的情况下,所述胶带的实用性受限。
10.在这些和其它情况下,阳离子固化的环氧体系是尤其合适的,并且在此特别是基于脂(环)族环氧化物的那些,其经由借助于成酸引发剂通过热(“热酸产生剂”,tag)和/或在紫外辐射的作用下(“光酸产生剂”,pag)的活化来反应。与缩水甘油醚相比,脂(环)族环氧化物可用这些引发剂更有效地固化(j.v.crivello,j.polym.sci.a polym.chem.,1999,37,4241-54)。然而,对于特别适合于这种活化的基于脂(环)族环氧化物的配制物,如在基于缩水甘油醚的反应性树脂的情况中那样,本领域技术人员无法获得如此丰富的不同的反应性树脂。可用的基于脂(环)族环氧化物的反应性树脂另外地具有低分子量,这可导致在热压制条件下或根据配制物甚至在室温下在一定程度上挤出的问题,因为尚未转化的低分子量的反应性树脂作为增塑剂起作用。因此,正在寻求的是用于胶粘剂体系、特别地用于膜形式的那些的反应性树脂,其不具有所述缺点或具有减少形式的所述缺点。
11.现有技术
12.wo 98/21287 a1描述了用于可热固化胶粘剂体系的可辐射固化的前体,其包括:(a)可辐射固化的单体/预聚物浆料,其应特别地被认为是聚(甲基)丙烯酸酯组分,(b)环氧树脂组分,(c)光引发剂组分和(d)亲核热活化剂。低聚物和聚合物环氧化物可用作组分(b)。未明确提及用脂环族环氧化物官能化的(共)聚合物。脂环族环氧化物甚至从根本上被描述为是不怎么有利的;参见所讨论文献的第19页第2行。未提出借助于tag或pag的固化。
13.us 4,552,604 a是“双固化”体系的另一实例,其中聚(甲基)丙烯酸酯是在环氧树脂存在下通过光聚合形成的。液体组合物的光聚合在衬垫上进行。最后使光聚合的膜热固化以用于粘合。利用光聚合以形成用于可热固化的环氧组分的聚合物基体。没有提及用形成超酸的引发剂固化。
14.ep 2 545 132 a1公开了基于聚丙烯酸酯的可光固化的压敏胶粘剂。聚丙烯酸酯含有小比例的环氧官能化的共聚单体,经由该共聚单体可使压敏胶粘剂交联。环氧官能化的共聚单体的比例如此之小,使得不可直接使用所述共聚物来获得具有远高于通过压敏胶粘剂获得的粘合强度的粘合强度的反应性胶带。
15.ep 819 746 a1描述了可固化胶粘膜,其配制物包括高分子量的聚丙烯酸酯、可光聚合的环氧组分和阳离子光引发剂。根据描述,聚丙烯酸酯同样可含有小比例、例如约2%的具有环氧基团的共聚物。关于环氧组分没有做出特定的选择。
16.ep 914 027 a1同样描述了可固化胶粘膜,其可含有聚丙烯酸酯、环氧组分和潜在固化剂。聚丙烯酸酯可含有小比例的(甲基)丙烯酸缩水甘油酯。
17.wo 2013/101693 a1公开了由借助于光引发而自由基聚合的丙烯酸酯单体混合物以及环氧组分制造的可热固化胶粘膜。没有提及环氧官能化的(甲基)丙烯酸酯。
18.wo 2015/048012 a1描述了可热固化的压敏胶粘剂体系,其包括可与苯并嗪反应的聚甲基丙烯酸酯组分。为此,它尤其可含有环氧基团,优选地经由甲基丙烯酸缩水甘油酯作为共聚单体引入聚合物中。该描述包括经由fox方程(u.w.gedde,polymer physics,
1999,kluwer,dordrecht,p.70)计算的玻璃化转变温度。fox方程允许理论估计均匀混合物的玻璃化转变温度,并且为此利用混合物的起始组分的玻璃化转变温度,其由混合物中这些组分的相应比例加权。其中使用的基础数据基于相应共聚单体的假想均聚物的玻璃化转变温度。为此,可使用对于具有非常高分子量的均聚物(即其中在玻璃化转变温度方面不随分子量有变化的那些)列出的列表值。所述fox方程不应与描述聚合物分子量对玻璃化转变温度的影响的fox-flory关系(方程g1)相混淆。因此,对于wo 2015/048012 a1中描述的聚合物,可认为是非常高的分子量,并且显然没有考虑使用具有较低分子量的聚合物。
19.wo 1999/057216 a1公开了包括20重量%-80重量%乙烯-乙酸乙烯酯共聚物和20重量%-80重量%也可为聚合物的环氧组分的配制物。引用的具体实例是含有甲基丙烯酸缩水甘油酯的聚合物。没有提及基于被脂(环)族环氧化物取代的(甲基)丙烯酸酯的聚合物。
20.wo 2012/165259 a1描述了可聚合液体胶粘剂配制物,通过uv辐射使其固化。为此,配制物含有具有脂环族环氧基团和(甲基)丙烯酸酯基团的单体。另外,配制物一方面含有用于(甲基)丙烯酸酯基团的自由基聚合的光引发剂以及另一方面含有用于环氧基团的阳离子聚合的光引发剂。然后照射同时引发两个反应过程。配制物具有对于液体胶粘剂体系来说典型的缺点,例如气味和在施用时的挤出倾向。此外,自由基固化操作可为不利的,因为这种类型的固化仅在照射期间进行,并且经由该机制的固化反应在阴影区域中没有到达胶粘剂。
技术实现要素:
21.目的
22.仍然需要如下的用于可固化胶粘剂组合物、特别地用于反应性胶带的反应性树脂:其可经由活化形成高粘合强度并且在活化步骤中具有最低的挤出倾向性,但同时在压制条件下仍具有非常好的适应特性并因此允许高粘合强度。为了以最佳的方式平衡这些相反的需求,因此正在寻求的是具有特别平衡的粘弹性特性的胶粘剂体系。
23.技术方案
24.该目的是通过如下的反应性树脂实现的:其基于环氧官能化(共)聚合物,所述环氧官能化(共)聚合物具有来自高于最小分子量且低于最大分子量的本发明的分子量范围的分子量。高于最小分子量的本发明的分子量范围包括其中未固化的环氧官能化(共)聚合物的玻璃化转变温度取决于分子量的分子量范围。聚合物分子量m和玻璃化转变温度tg之间的依赖性本身是已知的并且大致通过fox-flory关系来描述:
25.1/tg=1/t
g∞
+const/m
ꢀꢀꢀ
(g1)
26.其中t
g∞
是聚合物的玻璃化转变温度,在该温度下tg不再随分子量变化,且const是聚合物类型依赖性常数(t.fox,p.j.flory,j.polym.sci.,1954,14,315-319)。已经发现,在该分子量范围内环氧官能化(共)聚合物在其未固化状态下具有对于压制过程优异的粘弹性特性或在基于其的反应性胶粘剂配制物中导致对于压制过程优异的粘弹性特性、具体地导致特别有利的良好的适应特性和降低的挤出特性的平衡(对于该情况,压制温度高于玻璃化转变温度)。对于固化的反应性胶粘剂组合物,反应性另外允许非常好的粘合强度。由于玻璃化转变温度随在本发明的分子量范围内的分子量变化,因此在固化反应期间玻璃
化转变温度升高,这导致反应性胶粘剂体系的内部强度的升高和具有载荷承受能力的粘合状态。
27.因此,本发明涉及环氧官能化(共)聚合物、特别地可热固化和/或可辐射化学固化的那些,即具有在5000g/mol至200 000g/mol范围内的重均分子量的环氧官能化(共)聚(甲基)丙烯酸酯,基于超过30重量%至100重量%、优选地50重量%至100重量%(基于环氧官能化(共)聚合物所基于的单体(基于环氧官能化(共)聚合物的母体单体)的整体)的至少一种类型的用环氧基官能化的(甲基)丙烯酸类(共聚)单体(a)。
28.因此,本发明的聚合物是能通过至少一种单体、即一种或多种(甲基)丙烯酸酯单体和任选地另外地乙烯基类共聚单体的自由基聚合获得的聚合物。
29.分子量数值涉及通过测试方法b的借助于gpc的测量。
30.本发明包括如下内容:
31.实施方式1.聚合物,其能通过至少一种单体、即一种或多种(甲基)丙烯酸酯单体和任选地另外地乙烯基类共聚单体的自由基聚合获得,
[0032]-其中聚合物具有至少5000g/mol且至多200 000g/mol的分子量mw[0033]-且单体的至少一种用至少一个环氧基官能化,
[0034]
其特征在于
[0035]
环氧官能化的单体(a)的比例超过30重量%。
[0036]
实施方式2.如实施方式1所述的聚合物,其特征在于分子量mw为至少10 000g/mol、优选地至少20 000g/mol。
[0037]
实施方式3.如实施方式1或2所述的聚合物,其特征在于分子量mw为至多150 000g/mol、优选地至多100 000g/mol。
[0038]
实施方式4.如前述实施方式任一项所述的聚合物,其特征在于在环氧官能化的单体的至少一部分中的环氧基的所有或一些氧原子桥接脂族c-c键(脂族环氧基)。
[0039]
实施方式5.如前述实施方式任一项所述的聚合物,其特征在于在环氧官能化的单体的至少一部分中的环氧基的所有或一些氧原子桥接c-c键,所述c-c键为任选地杂取代的脂族烃环的一部分(脂环族环氧基)。
[0040]
实施方式6.如前述实施方式任一项所述的聚合物,其特征在于其在未交联状态下的玻璃化转变温度高于0℃、优选地高于25℃、非常优选地高于35℃。
[0041]
实施方式7.如前述实施方式任一项所述的聚合物,其特征在于其在未交联状态下的玻璃化转变温度低于120℃、优选地低于100℃、非常优选地低于80℃。
[0042]
实施方式8.如前述实施方式任一项所述的聚合物,其特征在于单体的至少一种为如下的硬单体,由该单体形成的均聚物在不取决于分子量的范围内具有至少25℃、特别地至少50℃的玻璃化转变温度。
[0043]
实施方式9.如前述实施方式任一项所述的聚合物,其特征在于单体的至少一种为如下的软单体,由该单体形成的均聚物在不取决于分子量的范围内具有低于25℃、特别地至多0℃的玻璃化转变温度。
[0044]
实施方式10.如前述实施方式任一项所述的聚合物,其特征在于单体的至少一种具有一种或多种其它官能团,即不为环氧基的这样的官能团。
[0045]
实施方式11.如实施方式10所述的聚合物,其特征在于其它官能团的至少一种为
含硅的基团。
[0046]
实施方式12.如实施方式10和11任一项所述的聚合物,其特征在于具有其它官能团的单体的比例为最高达10重量%、优选地最高达5重量%。
[0047]
实施方式13.如实施方式2所述的聚合物,其特征在于在固化状态下的聚合物的玻璃化转变温度比在未交联状态下的聚合物的玻璃化转变温度高至少40℃、优选地高至少100℃。
[0048]
实施方式14.反应性胶粘剂组合物,其包括如前述实施方式任一项所述的聚合物和至少一种用于涉及该聚合物的固化反应的引发剂。
[0049]
实施方式15.如实施方式14所述的反应性胶粘剂组合物,其特征在于在固化状态下的胶粘剂组合物的玻璃化转变温度比在未交联状态下的胶粘剂组合物的玻璃化转变温度高至少40℃、优选地高至少100℃。
附图说明
[0050]
图1a和1b显示渗出测试的程序。
[0051]
图2显示玻璃化转变温度的评价方式。
具体实施方式
[0052]
在本发明的上下文中,术语“(共)聚合物”共同用于均聚物或共聚物。在该文献的上下文中提及聚合物的情况下,这意指(共)聚合物,除非从相应的上下文以另外的方式明晰。
[0053]
在本发明的上下文中,术语“(共)聚(甲基)丙烯酸酯”理解为是指聚丙烯酸酯和聚甲基丙烯酸酯均聚物或者由(甲基)丙烯酸类单体和任何其它可共聚的共聚单体组成的聚丙烯酸酯和聚甲基丙烯酸酯共聚物。
[0054]
术语“(甲基)丙烯酸酯”和形容词“(甲基)丙烯酸类”共同是指来自以下的组的化合物:丙烯酸衍生物如特别地丙烯酸酯、以及甲基丙烯酸衍生物如特别地甲基丙烯酸酯。
[0055]
在本发明的上下文中,“可(共)聚合的”涉及一种类型的单体或至少两种类型的单体的混合物通过分子量增加反应形成(共)聚合物的能力。
[0056]
在优选的方式中,用至少一个环氧基官能化的(共聚)单体(a)的重均分子量为至少10 000g/mol、非常优选地至少20 000g/mol。进一步优选地,用至少一个环氧基官能化的(共聚)单体(a)的重均分子量为至多150 000g/mol、非常优选地至多100 000g/mol。
[0057]
根据环氧官能化(共)聚合物所基于的单体整体中的比例,用环氧基官能化的(甲基)丙烯酸类(共聚)单体(a)具有超过30重量%至100重量%、优选地至少50重量%的在环氧官能化(共)聚合物中的(共聚)单体含量。
[0058]
在非常优选的方式中,所使用的用环氧基官能化的(甲基)丙烯酸类(共聚)单体(a)为脂环族环氧化物,或者当存在两种或更多种用环氧基官能化的(甲基)丙烯酸类(共聚)单体(a)时,脂环族环氧化物用于一种、超过一种或所有这些用环氧基官能化的(甲基)丙烯酸类(共聚)单体(a)。特别有利地,脂环族环氧化物用于超过50重量%的(共聚)单体(a);更优选地,对于(共聚)单体(a)仅使用脂环族环氧化物。
[0059]
所述至少一种类型的环氧官能化(共)聚合物可任选地包括可源自以下单体的单
元(在该情况下至少存在共聚物),其中在下文中提及的单体类型(b)、(c)和(d)各自可存在而不论是否存在相应的其它类型的单体:
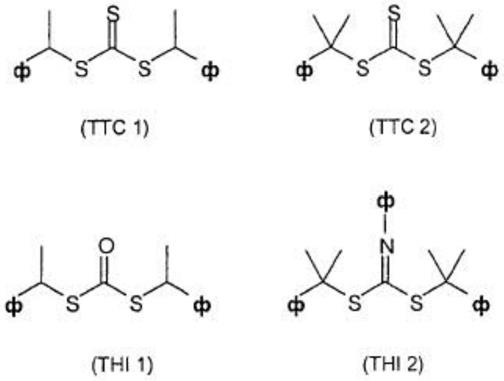
[0060]
(b)一种或多种类型的具有至少25℃、特别地至少50℃的玻璃化转变温度的共聚单体,其具有0重量%至小于70重量%、优选地0重量%至至多50重量%的共聚物中的共聚单体含量,
[0061]
和/或
[0062]
(c)一种或多种类型的具有低于25℃、特别地至多0℃的玻璃化转变温度的共聚单体,其具有0重量%至小于70重量%、优选地0重量%至至多50重量%的共聚物中的共聚单体含量,
[0063]
和/或
[0064]
(d)一种或多种类型的具有至少一种除环氧基以外的官能团、特别地含硅基团的共聚单体,其具有0重量%至10重量%、优选地0重量%至5重量%的共聚物中的共聚单体含量。
[0065]
在本文献的上下文中,(共)聚合物中的单体含量或(共聚)单体含量是指所讨论的(共)聚合物中的可溯源于这些(共聚)单体的重复单元(构建单元)的比例。因此,有利地选择用于制备相应共聚物的待聚合的聚合物混合物中的单体含量。
[0066]
本文献中的玻璃化转变温度数值涉及根据方法c的借助于dsc的测量。
[0067]
环氧官能化(共)聚合物的玻璃化转变温度优选地为至少0℃、非常优选地至少25℃、甚至更优选地至少35℃。它优选地为至多100℃、更优选地至多80℃。在本发明的替代实施方式中,官能化(共)聚合物(a)的玻璃化转变温度也可低于0℃。
[0068]
在作为胶粘剂体系使用的情况下或在胶粘剂体系中,在于加热和压制下施用时,胶粘剂体系软化,其润湿特性增加,并由此它可与待粘合基底形成接触。就此而言,本发明的环氧官能化(共)聚合物的分子量具有重要意义,因为对于给定的组合物,它影响熔体的粘弹性性质并且在此特别地熔体粘度。分子量越高,作为临时交联点的缠结对粘弹性特性的影响越明显。如果本发明的官能化(共)聚合物的分子量低于其缠结分子量,则相应聚合物在压制条件(即,高于玻璃化转变温度)下是极具流动性的,并且存在显著挤出的风险。相反,如果分子量过高,即在玻璃化转变温度不再随分子量变化的分子量范围内,则聚合物已过于显著地缠结,由此降低流动特性,使得在压制条件下不再确保良好的适应性。
[0069]
另外,本发明的环氧官能化(共)聚合物提供另外的优点。这是因为本发明利用如下发现:在其中可发生挤出的粘合步骤中,反应性体系通过经由升高的温度和/或(uv)辐射的活化而经历分子量的增加。在此发生两个过程:链增长和交联。这两个过程都是动力学控制的并且需要时间。如果在粘合条件下使用热量,则体系的粘度根据其温度依赖性而降低,这可导致挤出。如果分子量没有足够快地增加,作为粘度的分子量依赖性(其原则上抵消粘度的温度依赖性)的结果,无法补偿由于引入热量而引起的粘度下降。结果是不希望的胶粘剂材料的挤出和不完美的粘合结果。
[0070]
然而,本发明的聚合物已经具有基础分子量,因此链增长步骤已经在活化之前进行,并且仅需要进行交联以形成内聚性。
[0071]
(共聚)单体
[0072]
以下类型(a)、(b)、(c)和(d)的(共聚)单体在本发明的上下文中是有利的:
[0073]
(共聚)单体(a)
[0074]
用于(共聚)单体(a)的单体为式(i)的那些
[0075][0076]
其中
–
r1为
–
h或
–
ch3,
–
x
–
为
–
n(r3)
–
或
–o–
,
–
r3为
–
h或
–
ch3且
–
r2为环氧官能化的(杂)烃基。
[0077]
进一步优选地,r2基团包括具有2至30个碳原子并已用环氧基官能化的线性、支化、环状或多环烃基。该组的特别优选的代表是3,4-环氧基环己基取代的单体,例如甲基丙烯酸3,4-环氧基环己基甲酯、丙烯酸3,4-环氧基环己基甲酯、甲基丙烯酸3,4-环氧基环己酯、丙烯酸3,4-环氧基环己酯。
[0078]
不那么优选的但为本发明所涵盖的(共聚)单体(a)是含有缩水甘油基的(共聚)单体,例如丙烯酸缩水甘油酯或甲基丙烯酸缩水甘油酯。然而,缩水甘油基中的环氧乙烷单元的固化性质与脂(环)族环氧化物的固化性质不同,特别是在阳离子固化的情况下。此外,基于缩水甘油醚的体系出于涉及制备它们的工艺的原因而通常含有残留的卤素。然而,基于本发明的环氧官能化(共)聚合物的胶粘剂组合物优选地具有非常低的卤素含量(《1000ppm、优选地甚至《100ppm)、特别地基于氯和溴。
[0079]
(共聚)单体(a)的比例可用于调节(共)聚合物的包括机械性质在内的性质,并且还可用于调节含有这些聚合物的配制物的包括机械性质在内的性质,因为(共)聚合物中环氧基的比例在固化期间可用于调节交联密度。低的(共聚)单体(a)的比例导致弹性较小的胶粘剂层,较高的比例导致更显著弹性的胶粘剂层,各自相对于粘弹性性质。
[0080]
(共聚)单体(a)原则上可在其玻璃化转变温度方面自由地选择,其中特别有利地选择(共聚)单体(a),使得所得(共)聚合物的玻璃化转变温度根据所需的性质(例如压敏胶粘性的或非压敏胶粘性的)而在相应要求的范围内(另见下文)。
[0081]
在有利的方式中,可例如以这样的方式选择单体(a),使得对于其主要的数量、特别地对于所有单体(a)如下是适用的:由相应单体形成的假想均聚物的玻璃化转变温度(就此而言意指的是在不取决于分子量的玻璃化转变温度范围内的相应单体的均聚物的玻璃化转变温度,t
g∞
)为至少25℃、特别地至少50℃。
[0082]
在另一有利的方式中,可例如以这样的方式选择单体(a),使得对于其主要的数量、特别地对于所有单体(a)如下是适用的:由相应单体形成的假想均聚物的玻璃化转变温度(就此而言意指的是在不取决于分子量的玻璃化转变温度范围内的相应单体的均聚物的玻璃化转变温度,t
g∞
)为低于25℃、特别地低于0℃。
[0083]
共聚单体(b)
[0084]
共聚单体(b)特别地不具有环氧基。在共聚单体(b)范围内的可用共聚单体是本领域技术人员已知的所有(甲基)丙烯酸酯单体(特别地不含环氧基的那些)以及如下的其它可共聚的乙烯基单体,该乙烯基单体可与(共聚)单体(a)和任选地(c)和/或(d)共聚并且具有至少25℃、特别地至少50℃的作为假想均聚物的玻璃化转变温度(就此而言,这意味着在不取决于分子量的玻璃化转变温度范围内的相应单体的均聚物的玻璃化转变温度,t
g∞
)。
在本文献的上下文中,这种单体也称为“硬单体”。对于这样的共聚单体的选择可考虑的来源的实例是polymer handbook(j.brandrup,e.h.immergut,e.a.grulke(eds.),第4版,1999,j.wiley,hoboken,第1卷,第vi/193章)。根据wo 2015/082143 a1也可有利地使用的是所谓的大分子单体。优选这样的共聚单体,其基本上凭借其化学设计而在引发固化反应之前不具有与(共聚)单体(a)的环氧官能团的反应性或者具有与环氧官能团的任何反应相关的引发或催化作用,或者其与环氧官能团的反应性以另外的方式得到抑制。(共聚)单体(b)的比例可用于调节胶粘剂组合物的胶粘性性质和机械性质。它们具有使胶粘剂更硬的趋向性。
[0085]
共聚单体(c)
[0086]
共聚单体(c)特别地不具有环氧基。在共聚单体(c)范围内的可用共聚单体是本领域技术人员已知的所有(甲基)丙烯酸酯单体(特别地不含环氧基的那些)以及如下的其它可共聚的乙烯基单体,该乙烯基单体可与(共聚)单体(a)和存在的任何共聚单体(b)和/或(d)共聚并且具有低于25℃、特别地至多0℃的作为假想均聚物的玻璃化转变温度(就此而言,这意味着在不取决于分子量的玻璃化转变温度范围内的相应单体的均聚物的玻璃化转变温度,t
g∞
)。在本文献的上下文中,这种单体也称为“软单体”。对于这样的共聚单体的选择可考虑的来源的实例是polymer handbook(j.brandrup,e.h.immergut,e.a.grulke(eds.),第4版,1999,j.wiley,hoboken,第1卷,第vi/193章)。根据wo 2015/082143 a1也可有利地使用的是所谓的大分子单体。优选这样的共聚单体,其凭借其化学设计而在引发固化反应之前基本上不起与环氧官能团的任何反应相关的引发或催化作用、特别地不具有与(共聚)单体(a)的环氧官能团的反应性和/或其与环氧官能团的反应性已经以一些其它的方式受到抑制。共聚单体(c)的比例可用于调节胶粘剂的胶粘性性质和机械性质。它们具有使胶粘剂更软的趋向性,并且可实现压敏胶粘性。
[0087]
共聚单体(d)
[0088]
在共聚单体(d)范围内使用的单体特别地为可与(共聚)单体(a)和存在的任何共聚单体(b)和/或(c)共聚并且优化本发明的共聚物的胶粘性性质的那些。就此而言,作为有利的共聚单体,应特别地提及含硅的共聚单体和在此的含丙烯酸酯化烷氧基硅烷或甲基丙烯酸酯化烷氧基硅烷的共聚单体。实例为甲基丙烯酸3-(三乙氧基甲硅烷基)丙酯、丙烯酸3-(三乙氧基甲硅烷基)丙酯、丙烯酸3-(三甲氧基甲硅烷基)丙酯、甲基丙烯酸3-(三甲氧基甲硅烷基)丙酯、甲基丙烯酰氧基甲基三乙氧基硅烷、(甲基丙烯酰氧基甲基)三甲氧基硅烷、(3-丙烯酰氧基丙基)甲基二甲氧基硅烷、(甲基丙烯酰氧基甲基)甲基二甲氧基硅烷、γ-甲基丙烯酰氧基丙基甲基二甲氧基硅烷、甲基丙烯酰氧基丙基甲基二乙氧基硅烷、甲基丙烯酸3-(二甲氧基甲基甲硅烷基)丙酯、甲基丙烯酰氧基丙基二甲基乙氧基硅烷、甲基丙烯酰氧基丙基二甲基甲氧基硅烷。在上述化合物当中,特别优选甲基丙烯酸3-(三乙氧基甲硅烷基)丙酯、丙烯酸3-(三乙氧基甲硅烷基)丙酯、丙烯酸3-(三甲氧基甲硅烷基)丙酯和甲基丙烯酸3-(三甲氧基甲硅烷基)丙酯。
[0089]
共聚单体(d)也优选地不具有环氧基。
[0090]
制备
[0091]
环氧官能化(共)聚合物的制备通过母体(共聚)单体的(共)聚合实现并且可在物质中(以本体)、在一种或多种有机溶剂存在下、在水存在下或在有机溶剂和水的混合物中
进行。在此目的是保持尽可能少的使用的溶剂量。合适的有机溶剂是纯的烷烃(例如己烷、庚烷、辛烷、异辛烷、异己烷、环己烷)、芳族烃(例如苯、甲苯、二甲苯)、酯(例如乙酸乙酯、乙酸丙酯、乙酸丁基或乙酸己酯)、卤代烃(例如氯苯)、烷醇(例如甲醇、乙醇、乙二醇、乙二醇单甲醚)、酮(例如丙酮、丁酮)和醚(例如二乙醚、二丁醚)或其混合物。避免如下的化合物:其可在引发固化反应之前与环氧官能团反应或可引发或催化环氧官能团的反应;或者以一些其它的方式抑制其与环氧官能团的反应性。
[0092]
含水聚合反应可掺入水混溶性或亲水性共溶剂以确保反应混合物在单体转化期间为均相形式。有利地,对于本发明可用的共溶剂选自以下:脂族醇、二醇、醚、二醇醚、聚乙二醇、聚丙二醇、酯、醇衍生物、羟基醚衍生物、酮等、及其衍生物和混合物。避免如下的化合物:其可与环氧官能团反应和/或可引发或催化环氧官能团的反应;或者以一些其它的方式被抑制其与环氧官能团的反应性。
[0093]
本发明的环氧官能化(共)聚合物有利地使用常规的自由基聚合或受控的自由基聚合来制备。对于自由基聚合,优选使用含有用于聚合的自由基引发剂(聚合引发剂)、特别地经历热分解而形成自由基的偶氮或过氧引发剂的引发剂体系。然而,惯常用于丙烯酸酯和/或甲基丙烯酸酯并且为本领域技术人员熟悉的所有聚合引发剂是合适的。c中心自由基的产生描述于houben-weyl,methoden der organischen chemie,vol.e 19a,p.60-147中。优选地,类似地使用这些方法。
[0094]
关于(共)聚合物的制备提及的自由基聚合引发剂不应与用于固化可固化胶粘剂组合物的活化剂相混淆。
[0095]
自由基源的实例是过氧化物、氢过氧化物和偶氮化合物。典型的自由基引发剂的一些非排他性实例包括过氧二硫酸钾、过氧化二苯甲酰、氢过氧化枯烯、过氧化环己酮、过氧化二叔丁基、偶氮双异丁腈、环己基磺酰基乙酰基过氧化物、过碳酸二异丙酯、过辛酸叔丁酯、苯频哪醇。特别优选使用2,2'-偶氮双(2-甲基丁腈)或2,2-偶氮双(2,4-二甲基戊腈)作为自由基聚合引发剂。
[0096]
根据温度和所需的转化率,聚合时间在4和72小时之间。可选择的反应温度越高,即反应混合物的热稳定性越高,可选择的反应时间越短。
[0097]
为了引发聚合,热量的输入对于经历热分解的聚合引发剂是必不可少的。对于经历热分解的聚合引发剂,根据引发剂类型,可通过加热至50℃或更高来引发聚合。优选不超过100℃、非常优选地不超过80℃的引发剂温度。
[0098]
在有利的程序中,使用氮氧自由基(氮氧化物)如(2,2,5,5-四甲基-1-吡咯烷基)氧基(proxyl)、(2,2,6,6-四甲基-1-哌啶基)氧基(tempo)、proxyl或tempo的衍生物以及本领域技术人员熟悉的其它氮氧自由基来稳定自由基。
[0099]
在替代的程序中可根据其制备环氧官能化(共)聚合物的许多其它聚合方法可选自现有技术:wo 96/24620 a1描述了其中使用非常特定的自由基化合物如基于咪唑烷的含磷氮氧自由基的聚合方法。wo 98/44008 a1公开了特定的基于吗啉、哌嗪酮和哌嗪二酮的氮氧基。de 199 49 352 a1描述了杂环烷氧基胺作为受控自由基聚合中的调节剂。
[0100]
可使用的其它受控聚合方法是原子转移自由基聚合(atrp),其中所用的聚合引发剂优选为单官能或双官能的仲或叔卤化物,并且使用cu、ni、fe、pd、pt、ru、os、rh、co、ir、ag或au的络合物提取卤素。atrp的不同选项还描述于文献us 5,945,491 a、us 5,854,364 a
和us 5,789,487 a中。
[0101]
进行的其它制备方法是raft聚合(可逆加成-断裂链转移聚合)的变体。例如,在文献wo 98/01478 a1和wo 99/31144 a1中详述了聚合方法。对于制备特别有利的是通式结构r”'-s-c(s)-s-r”'的三硫代碳酸酯(macromolecules,2000,33,243-245)。
[0102]
在非常有利的变体中,例如,三硫代碳酸酯(ttc1)和(ttc2)或硫代化合物(thi1)和(thi2)用于聚合,其中φ是苯环、氰基或者饱和或不饱和脂族基团,所述苯环可为未官能化的或者通过直接地或经由酯或醚桥键合的烷基或芳基取代基官能化的。苯环φ可任选地具有一个或多个聚合物嵌段,例如聚丁二烯、聚异戊二烯或聚苯乙烯,仅举几例。例如,官能化可为卤素、羟基、环氧基,然而该列表对完备性不做任何要求。
[0103][0104]
关于上述受控的自由基聚合,优选如下的的聚合引发剂体系,其含有用于聚合的自由基聚合引发剂、特别地上面已经列举的经历热分解而形成自由基的偶氮或过氧引发剂。然而,所有已知用于丙烯酸酯和/或甲基丙烯酸酯的聚合引发剂适合于该目的。此外,还可使用仅在uv照射下释放自由基的自由基源。至关重要的是,这些引发剂不可活化环氧官能团的任何反应。
[0105]
分子量调节的目的也可使用根据现有技术的链转移剂完成,条件是它们对环氧基团没有任何反应性或者它们与环氧基团的反应性已经以一些其它的方式得到抑制。
[0106]
所需的分子量优选地通过聚合方法建立,无论它们是受控聚合方法还是非受控聚合方法,其中不使用如下的试剂:其在引发胶粘膜的固化反应之前可与环氧官能团反应或可引发或催化环氧官能团的反应;或者它们与环氧官能团的反应性已经以一些其它的方式得到抑制。
[0107]
所需分子量的建立另外地且更优选地可经由聚合引发剂与(共聚)单体的使用比率和/或(共聚)单体的浓度来实现。
[0108]
基于本发明的环氧官能化(共)聚合物的反应性胶粘剂体系
[0109]
本发明的环氧官能化(共)聚合物具有优异的与潜在反应性引发剂(固化剂)(a)组合的适合性,所述引发剂可热活化和/或辐射化学(特别地使用uv辐射)活化,并且于是可有利地用作可固化反应性胶粘剂体系或用于可固化反应性胶粘剂体系中。
[0110]
相应地,本发明还涉及可固化反应性胶粘剂体系,其包括或包含至少一种类型的本发明的环氧官能化(共)聚合物和至少一种类型的潜在反应性引发剂。借助于这样的可固
化反应性胶粘剂体系,则可产生由以下组成的胶粘复合物:待粘合的第一基底、待粘合的第二基底和包括至少一种类型的本发明的可固化反应性胶粘剂体系的粘合产品。当可固化反应性胶粘剂体系在复合物中为固化形式时获得胶粘复合物的粘合强度。
[0111]
在本发明的上下文中,命名为“经固化的体系”或“经固化的胶粘剂组合物(物质)”是指具有官能化(共)聚合物的胶粘剂组合物已经经由固化剂组分的作用和任选地额外刺激(如热和/或辐射)而被活化,并且涉及(共)聚合物的环氧基的反应已经发生。然而,不需要可化学上参与固化反应的所有环氧基都已反应。相反,官能团的50%的转化率已经可带来足够高的玻璃化转变温度并且对于粘合应用具有非常好的适合性。在此以举例的方式提到50%的转化率。所做的说明也可适用于更高的转化率如60%、70%、80%、90%或100%、或甚至更低的转化率如40%或30%。重要的是,在进行固化后,粘合性质符合应用;换言之,例如,根据测试e的耐推出性为至少2.0n/mm2。
[0112]
因此,本发明的反应性胶粘剂体系有利地含有至少一种类型的固化剂。然而,固化剂不是绝对必要的,因为本发明的(共)聚合物在足够的温度下是自交联的。然而,如果使用固化剂,则选择它们,使得所得配制物在其反应性方面具有非常显著的潜伏性。这意味着胶粘剂体系或基于其的胶粘膜在特定条件下(例如在室温或甚至略微升高的温度如35℃或甚至50℃下和/或在排除光的情况下)显示基本上没有反应或甚至显示完全没有反应。反应理想地首先遵循可由升高的温度和/或(uv)辐射触发的活化刺激。在本发明的上下文中,经由可借助于dsc实验(测试d)测定的活化温度定义可热活化体系的潜伏性。由此确定的本发明的热固化剂的活化温度为至少60℃、优选地至少75℃、非常优选地至少90℃。它为至多150℃、优选地至多120℃。在可以(uv)辐射化学的方式活化的固化剂的情况下,典型地存在热潜伏性。相反,在此必须确保的是排除光直至所需的活化时间点。
[0113]
合适的固化剂的实例包括可热活化的酸形成剂tag。热的影响导致由引发剂物质形成称为超酸的强酸,并且该酸可引起环氧基的开环。在本发明的上下文中可用于环氧基的阳离子固化的可热活化引发剂特别地为吡啶鎓盐、铵盐(特别地苯胺鎓盐)和硫鎓盐(特别地四氢噻吩鎓盐)、以及镧系元素三氟甲磺酸盐。
[0114]
n-苄基吡啶鎓盐和苄基吡啶鎓盐是非常有利的,其中芳族体系可为例如被烷基、烷氧基、卤素或氰基取代的。
[0115]
j.polym.sci.a,1995,33,505ff、us 2014/0367670 a1、us 5,242,715,j.polym.sci.b,2001,39,2397ff、ep 393893a1、macromolecules,1990,23,431ff、macromolecules,1991,24,2689、macromol.chem.phys.,2001,202,2554ff、wo 2013/156509 a2和jp 2014/062057 a1提到了在本发明的本上下文中可使用的相应化合物。
[0116]
在可商购获得的引发剂体系中,非常有利地可使用的化合物的实例包括来自sanshin的san-aid si 80l、san-aid si 100l、san-aid si 110l、san-aid si b2a、san-aid si b3、san-aid si b3a和san-aid si b4,来自adeka的opton cp-66和opton cp-77,以及来自king industries的k-pure tag 2678、k-pure cxc 1612和k-pure cxc 1614、k-pure cxc 1615、k-pure cxc 1821。
[0117]
另外地镧系元素三氟甲磺酸盐是可用的,如三氟甲磺酸钐(iii)、三氟甲磺酸镱(iii)、三氟甲磺酸铒(iii)或三氟甲磺酸镝(iii)(可得自sigma aldrich)、和三氟甲磺酸镧(iii)(可得自alfa aesar)。
[0118]
充当上述阳离子的抗衡离子的阴离子的实例包括四氟硼酸根、四苯基硼酸根、六氟磷酸根、高氯酸根、四氯高铁酸根、六氟砷酸根、六氟锑酸根、五氟羟基锑酸根、六氯锑酸根、四(五氟苯基)硼酸根、四(五氟甲基苯基)硼酸根、双(三氟甲磺酰基)酰胺和三(三氟甲磺酰基)甲烷化物。另外可用的是根据jp 2012-056915 a1和ep 393893 a1的阴离子。优选基本上不含氯和溴的引发剂。阴离子优选地为不含砷酸根的和不含锑酸根的。
[0119]
本领域技术人员知晓根据本发明同样可用的其它体系。用于阳离子固化的潜在反应性可热活化固化剂以未组合的形式或作为两种或更多种可热活化固化剂的组合使用。
[0120]
用于固化、特别地阳离子固化的可热活化固化剂的比例优选地为至少0.1重量%且至多5重量%、优选地至少0.3重量%且至多3重量%,基于总配制物。
[0121]
在本发明的上下文中有利的活化温度,即可引发官能化的(共)聚合物的阳离子固化的那些温度,为至少60℃、优选地至少75℃、进一步优选地至少90℃。在这些温度范围内固化/引发是优选的,以不热损坏热敏基底。对于耐热的基底,较高的固化温度也是可想到的,例如120℃、150℃、180℃、200℃或甚至更高,并且对于一些粘合目的甚至是优选的。固化时间在此可为15分钟或更长或2小时或更短,然而不排除明显更短(例如10秒、30秒、60秒、120秒、240秒、5分钟或10分钟)或甚至更长的固化时间。
[0122]
另外地,非常优选与本发明的环氧官能化(共)聚合物组合的是可以(uv)辐射化学的方式活化的酸形成剂pag。这些可经由uv引发而引起环氧基的阳离子固化反应。
[0123]
在用于阳离子uv固化的固化剂(引发剂)当中,特别地基于硫鎓、碘鎓和茂金属的体系为可使用的。
[0124]
对于基于硫鎓的阳离子的实例,参考us 6,908,722 b1(特别地第10-21栏)中给出的细节。
[0125]
充当上述阳离子的抗衡离子的阴离子的实例包括四氟硼酸根、四苯基硼酸根、六氟磷酸根、高氯酸根、四氯高铁酸根、六氟砷酸根、六氟锑酸根、五氟羟基锑酸根、六氯锑酸根、四(五氟苯基)硼酸根、四(五氟甲基苯基)硼酸根、双(三氟甲磺酰基)酰胺和三(三氟甲磺酰基)甲烷化物。可使用的其它阴离子、特别地对于基于碘鎓的固化剂(引发剂)还有氯根、溴根或碘根。另外可用的是根据jp 2012-056915 a1和ep 393893 a1的阴离子。在此也优选基本上不含氯和溴的引发剂。在此阴离子也优选地为不含砷的和不含锑的。
[0126]
更具体地,可使用的体系包括:
[0127]
·
硫鎓盐(参见例如us 4,231,951 a、us 4,256,828 a、us 4,058,401 a、us 4,138,255 a和us 2010/063221 a1)如三苯基硫鎓六氟砷酸盐、三苯基硫鎓六氟硼酸盐、三苯基硫鎓四氟硼酸盐、三苯基硫鎓四(五氟苄基)硼酸盐、甲基二苯基硫鎓四氟硼酸盐、甲基二苯基硫鎓四(五氟苄基)硼酸盐、二甲基苯基硫鎓六氟磷酸盐、三苯基硫鎓六氟磷酸盐、三苯基硫鎓六氟锑酸盐、二苯基萘基硫鎓六氟砷酸盐、三甲苯基硫鎓六氟磷酸盐、茴香基二苯基硫鎓六氟锑酸盐、4-丁氧基苯基二苯基硫鎓四氟硼酸盐、4-丁氧基苯基二苯基硫鎓四(五氟苄基)硼酸盐、4-氯苯基二苯基硫鎓六氟锑酸盐、三(4-苯氧基苯基)硫鎓六氟磷酸盐、二-(4-乙氧基苯基)甲基硫鎓六氟砷酸盐、4-乙酰基苯基二苯基硫鎓四氟硼酸盐、4-乙酰基苯基二苯基硫鎓四(五氟苄基)硼酸盐、三(4-硫代甲氧基苯基)硫鎓六氟磷酸盐、二(甲氧基磺酰基苯基)甲基硫鎓六氟锑酸盐、二(甲氧基萘基)甲基硫鎓四氟硼酸盐、二(甲氧基萘基)甲基硫鎓四(五氟苄基)硼酸盐、二(甲氧羰基苯基)甲基硫鎓六氟磷酸盐、(4-辛氧基苯基)二
苯基硫鎓四(3,5-双三氟甲基苯基)硼酸盐、三[4-(4-乙酰基苯基)硫代苯基]硫鎓四(五氟苯基)硼酸盐、三(十二烷基苯基)硫鎓四(3,5-双(三氟甲基)苯基)硼酸盐、4-乙酰氨基苯基二苯基硫鎓四氟硼酸盐、4-乙酰氨基苯基二苯基硫鎓四(五氟苄基)硼酸盐、二甲基萘基硫鎓六氟磷酸盐、三氟甲基二苯基硫鎓四氟硼酸盐、三氟甲基二苯基硫鎓四(五氟苄基)硼酸盐、苯基甲基苄基硫鎓六氟磷酸盐、5-甲基噻蒽鎓六氟磷酸盐、10-苯基-9,9-二甲基噻吨鎓六氟磷酸盐、10-苯基-9-氧代噻吨鎓四氟硼酸盐、10-苯基-9-氧代噻吨鎓四(五氟苄基)硼酸盐、5-甲基-10-氧代噻蒽鎓四氟硼酸盐、5-甲基-10-氧代噻蒽鎓四(五氟苄基)硼酸盐、和5-甲基-10,10-二氧代噻蒽鎓六氟磷酸盐,
[0128]
·
碘鎓盐(参见例如us 3,729,313 a、us 3,741,769 a、us 4,250,053 a、us 4,394,403 a和us 2010/063221 a1)如二苯基碘鎓四氟硼酸盐、
[0129]
二(4-甲基苯基)碘鎓四氟硼酸盐、
[0130]
苯基-4-甲基苯基碘鎓四氟硼酸盐、
[0131]
二(4-氯苯基)碘鎓六氟磷酸盐、二萘基碘鎓四氟硼酸盐、
[0132]
二(4-三氟甲基苯基)碘鎓四氟硼酸盐、二苯基碘鎓六氟磷酸盐、
[0133]
二(4-甲基苯基)碘鎓六氟磷酸盐、二苯基碘鎓六氟砷酸盐、
[0134]
二(4-苯氧基苯基)碘鎓四氟硼酸盐、
[0135]
苯基-2-噻吩基碘鎓六氟磷酸盐、
[0136]
3,5-二甲基吡唑基-4-苯基碘鎓六氟磷酸盐、二苯基碘鎓六氟锑酸盐、
[0137]
2,2'-二苯基碘鎓四氟硼酸盐、
[0138]
二(2,4-二氯苯基)碘鎓六氟磷酸盐、
[0139]
二(4-溴苯基)碘鎓六氟磷酸盐、
[0140]
二(4-甲氧基苯基)碘鎓六氟磷酸盐、
[0141]
二(3-羧基苯基)碘鎓六氟磷酸盐、
[0142]
二(3-甲氧基羰基苯基)碘鎓六氟磷酸盐、
[0143]
二(3-甲氧基磺酰基苯基)碘鎓六氟磷酸盐、
[0144]
二(4-乙酰氨基苯基)碘鎓六氟磷酸盐、
[0145]
二(2-苯并噻吩基)碘鎓六氟磷酸盐、
[0146]
二芳基碘鎓三三氟甲磺酰基甲基化物如
[0147]
二苯基碘鎓六氟锑酸盐、
[0148]
二芳基碘鎓四(五氟苯基)硼酸盐如
[0149]
二苯基碘鎓四(五氟苯基)硼酸盐、
[0150]
(4-n-二甲硅烷氧基(desiloxy)苯基)苯基碘鎓六氟锑酸盐、
[0151]
[4-(2-羟基-n-四二甲硅烷氧基)苯基]苯基碘鎓六氟锑酸盐、
[0152]
[4-(2-羟基-n-四二甲硅烷氧基)苯基]苯基碘鎓三氟磺酸盐、
[0153]
[4-(2-羟基-n-四二甲硅烷氧基)苯基]苯基碘鎓六氟磷酸盐、
[0154]
[4-(2-羟基-n-四二甲硅烷氧基)苯基]苯基碘鎓四(五氟苯基)硼酸盐、
[0155]
双(4-叔丁基苯基)碘鎓六氟锑酸盐、
[0156]
双(4-叔丁基苯基)碘鎓六氟磷酸盐、
[0157]
双(4-叔丁基苯基)碘鎓三氟磺酸盐、
[0158]
双(4-叔丁基苯基)碘鎓四氟硼酸盐、
[0159]
双(十二烷基苯基)碘鎓六氟锑酸盐、
[0160]
双(十二烷基苯基)碘鎓四氟硼酸盐、
[0161]
双(十二烷基苯基)碘鎓六氟磷酸盐、
[0162]
双(十二烷基苯基)碘鎓三氟甲磺酸盐、
[0163]
二(十二烷基苯基)碘鎓六氟锑酸盐、
[0164]
二(十二烷基苯基)碘鎓三氟甲磺酸盐、
[0165]
二苯基碘鎓硫酸氢盐、
[0166]
4,4'-二氯二苯基碘鎓硫酸氢盐、4,4'-二溴二苯基碘鎓硫酸氢盐、
[0167]
3,3'-二硝基二苯基碘鎓硫酸氢盐、4,4'-二甲基二苯基碘鎓硫酸氢盐、
[0168]
4,4'-双-琥珀酰亚氨基二苯基碘鎓硫酸氢盐、3-硝基二苯基碘鎓硫酸氢盐、4,4'-二甲氧基二苯基碘鎓硫酸氢盐
[0169]
双(十二烷基苯基)碘鎓四(五氟苯基)硼酸盐、
[0170]
(4-辛氧基苯基)苯基碘鎓四(3,5-双-三氟甲基苯基)硼酸盐和(甲苯基枯基)碘鎓四(五氟苯基)硼酸盐,以及
[0171]
·
二茂铁盐(参见例如ep 542 716b1)如η
5-(2,4-环戊二烯-1-基)-[(1,2,3,4,5,6,9)(1-甲基乙基)苯]铁。
[0172]
商业化光引发剂的实例包括来自union carbide的cyracure uvi-6990、cyracure uvi-6992、cyracure uvi-6974和cyracure uvi-6976,来自adeka的optomer sp-55、optomer sp-150、optomer sp-151、optomer sp-170和optomer sp-172,来自sanshin chemical的san-aid si-45l、san-aid si-60l、san-aid si-80l、san-aid si-100l、san-aid si-110l、san-aid si-150l和san-aid si-180l,来自sartomer的sarcat cd-1010、sarcat cd-1011和sarcat cd-1012,来自degussa的degacure k185,来自rhodia的rhodorsil photoinitiator 2074,来自nippon soda的ci-2481、ci-2624、ci-2639、ci-2064、ci-2734、ci-2855、ci-2823和ci-2758,来自igm resins的omnicat 320、omnicat 430、omnicat 432、omnicat 440、omnicat 445、omnicat 550、omnicat 550 bl和omnicat 650,来自daicel的daicat ii,来自daicel-cytec的uvac 1591,来自3m的ffc 509,来自midori kagaku的bbi-102、bbi-103、bbi-105、bbi-106、bbi-109、bbi-110、bbi-201、bbi-301、bi-105、dpi-105、dpi-106、dpi-109、dpi-201、dts-102、dts-103、dts-105、nds-103、nds-105、nds-155、nds-159、nds-165、tps-102、tps-103、tps-105、tps-106、tps-109、tps-1000、mds-103、mds-105、mds-109、mds-205、mpi-103、mpi-105、mpi-106、mpi-109、ds-100、ds-101、mbz-101、mbz-201、mbz-301、nai-100、nai-101、nai-105、nai-106、nai-109、nai-1002、nai-1003、nai-1004、nb-101、nb-201、ndi-101、ndi-105、ndi-106、ndi-109、pai-01、pai-101、pai-106、pai-1001、pi-105、pi-106、pi-109、pyr-100、si-101、si-105、si-106和si-109,来自nippon kayaku的kayacure pci-204、kayacure pci-205、kayacure pci-615、kayacure pci-625、kayarad 220和kayarad 620、pci-061t、pci-062t、pci-020t、pci-022t,来自sanwa chemical的ts-01和ts-91,来自deuteron的deuteron uv 1240,来自evonik的tego photocompound 1465n,来自ge bayer silicones的uv 9380 c-d1,来自cytec的fx 512,来自bluestar silicones的silicolease uv cata 211以及来自basf的
irgacure 250、irgacure 261、irgacure 270、irgacure pag 103、irgacure pag 121、irgacure pag 203、irgacure pag 290、irgacure cgi 725、irgacure cgi 1380、irgacure cgi 1907和irgacure gsid 26-1。
[0173]
本领域技术人员知晓根据本发明同样可用作固化剂的其它体系。光引发剂以未组合的形式或作为两种或更多种光引发剂的组合使用。
[0174]
在小于350nm处并且有利地在大于250nm处呈现吸收的光引发剂是有利的。同样可使用在高于350nm、例如在紫外光范围内吸收的引发剂。特别优选使用基于硫鎓的光引发剂,因为它们呈现有利的uv吸收特性。
[0175]
用于阳离子固化的可光化学活化的固化剂(引发剂)的比例优选地为至少0.1重量%且至多5重量%、优选地至少0.3重量%且至多3重量%,基于总组合物。
[0176]
选择可通过两种刺激方法(通过热和通过(uv)辐射)活化的固化剂也为有利的。也可使用可热活化和可光化学活化的固化剂(引发剂)的组合。
[0177]
即使优选与本发明的环氧官能化(共)聚合物组合的tag和pag体系,替代地或以组合的形式也可想到其它固化剂体系。在此还优选在配制物中具有潜伏性的体系。
[0178]
实例包括潜在反应性二胺或多官能胺、二羧酸或多官能羧酸、双官能酸酐或多官能酸酐、伯二硫醇或多官能伯硫醇。关于潜伏性的特别有利的反应配对物(共反应物)是在室温下为固体并且在非软化状态下不可溶于本发明的聚合物或含有所述聚合物的混合物中但在软化状态下可溶或两种熔体彼此混溶的那些。
[0179]
还可想到的是固化剂(引发剂),其为胶囊形式并且在热的影响下分布在膜基体中并且然后可导致反应。
[0180]
在未固化状态下的环氧官能化(共)聚合物具有低于如下温度的第一玻璃化转变温度:在该温度下由包括环氧官能化(共)聚合物的胶粘膜和待粘合基底组成的胶粘复合物通过层压产生。在本发明的上下文中,用于层压的温度称为“层压温度”。在该情况下未固化(共)聚合物的层压温度和玻璃化转变温度之间的温度差优选为至少20℃、特别地至少40℃,其中层压温度高于玻璃化转变温度。
[0181]
另外,当在未固化状态下的包括环氧官能化(共)聚合物的可固化反应性胶粘剂体系具有低于如下温度的第一玻璃化转变温度时则为有利的:在该温度下由反应性胶粘膜和待粘合基底组成的胶粘复合物通过层压产生,使得配制物在层压条件下在压力下允许在规定时间内对基底的充分润湿。层压温度和玻璃化转变温度之间的温度差优选为至少20℃、特别地至少40℃或甚至至少100℃,其中层压温度高于玻璃化转变温度。层压温度有利地在40℃和100℃之间、特别地在50℃和80℃之间。它低于活化温度,即如下的温度:如果胶粘剂体系是可热活化的,则在该温度下引发可固化胶粘剂组合物的固化。在该情况下,层压温度和活化温度之差有利地为至少20℃、特别地至少40℃。
[0182]
在以辐射化学的方式固化的胶粘剂体系中使用(uv)辐射,而非活化温度。这可在层压温度下(这是优选的)或在另外的温度下进行。
[0183]
优选地,环氧官能化(共)聚合物在标准条件下(23℃,50%相对湿度)不是压敏胶粘性的。在不完全反应的状态下,其具有至少0℃、优选地至少25℃的玻璃化转变温度。这可有利地使用,由此使包括该(共)聚合物的胶粘剂配制物不是压敏胶粘性的。该特性允许胶粘剂产品在粘合过程中的有利定位并且不在错误的位置中过早地粘至表面。此外,发现该
特性对于可热固化反应性胶粘剂体系是有利的,因为在玻璃/粘弹性状态下的任何反应性显著地(动力学地)降低并且由此实现改善的潜伏性。但是聚合物也可为略微压敏胶粘性的。
[0184]
胶粘剂组合物在标准条件下(23℃,50%相对空气湿度)可为压敏胶粘性的。在该情况下,其具有低于0℃、优选地至多-25℃的在未固化状态下的玻璃化转变温度。这些特性简化修整工艺如用于后续粘合过程的胶带部分的尺寸初定或者在胶粘剂产品结构的制造和部件粘合中的层压步骤。在层压过程中,在这种情况下以升高的温度加工并非绝对必要的,反而在室温下层压是可能的,因为胶粘剂组合物和待粘合基底之间的充分接触已经可经由层压压力实现。在该情况下uv辐射对于固化是优异的选择。然而,热活化也是可能的。
[0185]
术语“压敏胶粘剂”或“压敏胶粘剂组合物(物质)”(psa)通常被理解为是指那些粘弹性聚合物材料,其任选地经由合适地添加另外的组分(例如增粘树脂)而在使用温度(除非另有说明,否则在室温下,即23℃)下具有持久的粘性和永久的胶粘性并且在与多个表面接触时粘附并且特别地立即粘住(具有所谓的“粘性”[也称为胶粘粘性或解除粘性])。即使在使用温度下,它们也能够在不通过溶剂或通过加热活化的情况下(任选地在或大或小的压力的影响下)充分地润湿待粘合基底,使得在组合物和基底之间可形成足以造成粘附的相互作用。
[0186]
胶粘剂组合物在标准条件下(23℃,50%相对空气湿度)可仅具有低的或不具有压敏粘附性。为了建立这一点,它于是在未固化状态下具有至少0℃、优选地至少25℃的玻璃化转变温度。该特性允许胶粘剂产品在粘合过程中的有利定位并且不在错误的位置中过早地粘至表面。此外,发现该特性对于潜在反应性胶粘剂体系是有利的,因为在玻璃/粘弹性状态下的任何反应性显著地(动力学地)降低并且由此实现改善的潜伏性。对于层压过程,在该情况下,不仅需要压力,而且需要升高的温度。
[0187]
可如在反应性胶粘剂组合物中所述地使用环氧官能化(共)聚合物。除了固化剂(a)之外,有利地将它们与另外的成分组合,所述另外的成分根据需要调节胶粘剂体系的性质。就此而言应提及成膜剂(b)、增粘树脂(c)、低粘度反应性树脂(d)、填料(e)和粘附促进剂(f)和另外的掺混物/添加剂(g)。
[0188]
成膜剂(b)
[0189]
合适的用于本发明的反应性胶粘剂组合物的成膜剂是热塑性材料、弹性体和热塑性弹性体。特别地选择它们,使得它们在与另外的配制物成分组合的情况下可获得那些胶粘剂组合物:其一方面对于胶带制造商处已经另一方面对于胶带使用者在可加工性方面、在胶粘性性质方面和在胶粘膜的尺寸稳定性的进一步改善(例如关于胶粘剂产品的施用和在热层压工艺中的挤出特性)方面为有利的,仅举几个特别重要的要求。其比例典型地为至多75重量%或至多50重量%。也可摒弃这样的聚合物。
[0190]
在有利的程序中,热塑性材料用作基体聚合物(b)。实例是半结晶聚烯烃和乙烯-乙酸乙烯酯共聚物(eva)。优选的聚烯烃由乙烯、丙烯、丁烯和/或己烯制备,其中在各情况下可使纯的单体聚合或使所提及单体的混合物共聚。经由聚合方法和通过单体的选择可控制聚合物的物理和机械性质,例如软化温度和/或特定的机械性质。
[0191]
弹性体非常有利地可用作基体聚合物(b)。实例包括橡胶或合成橡胶作为胶粘剂组合物的起始材料。无论是对于来自天然橡胶或合成橡胶的橡胶,还是来自天然橡胶和/或
合成橡胶的任何共混物,在此有多种可能的变体,其中天然橡胶原则上可根据所需的纯度和粘度水平选自任何可用的品质,例如绉纱、rss、ads、tsr或cv类型,并且合成橡胶可选自无规共聚的丁苯橡胶(sbr)、丁二烯橡胶(br)、合成聚异戊二烯(ir)、丁基橡胶(iir)、卤化丁基橡胶(xiir)、丙烯酸酯橡胶(acm)、epdm、聚丁烯或聚异丁烯。弹性体也可为(部分)氢化的。
[0192]
腈橡胶是非常有利的,特别是已经热聚合的那些、以及具有在15%和50%之间、优选地在30%和45%之间的丙烯腈含量和在30和110之间、优选地在60和90之间的门尼粘度(ml 1+4,100℃)的那些。
[0193]
也非常有利的是由(共聚)单体(b)、(c)和/或(d)形成并且具有至少250 000g/mol且典型地至多5 000 000g/mol、特别地至少500 000g/mol且至多2 000 000g/mol的重均分子量的聚(甲基)丙烯酸酯。这些聚(甲基)丙烯酸酯的玻璃化转变温度可特别地低于25℃或甚至低于0℃、特别地低于-25℃。以此方式,压敏胶粘性反应性胶粘剂体系是可获得的。
[0194]
此外有利的是热塑性弹性体并且在此特别地嵌段共聚物、星形共聚物和/或接枝共聚物,其具有300 000g/mol或更小、优选地200 000g/mol或更小的(重均)分子量mw。较小的分子量在此由于其改善的可加工性而为优选的。分子量不应低于50 000g/mol。
[0195]
具体实例是苯乙烯-丁二烯嵌段共聚物(sbs)、苯乙烯-异戊二烯嵌段共聚物(sis)、苯乙烯-(异戊二烯/丁二烯)嵌段共聚物(sibs)和(部分)氢化变体如苯乙烯-(乙烯/丁烯)嵌段共聚物(sebs)、苯乙烯-(乙烯/丙烯)嵌段共聚物(seps、seeps)、苯乙烯-(丁烯/丁基)嵌段共聚物(sbbs)、苯乙烯-异丁烯嵌段共聚物(sibs)和聚甲基丙烯酸甲酯-聚丙烯酸酯嵌段共聚物。这些嵌段共聚物可以线性或多臂结构的形式、以二嵌段共聚物、三嵌段共聚物或多嵌段共聚物的形式、或以不同类型的混合物的形式使用。
[0196]
热塑性弹性体的另外的有利实例是热塑性聚氨酯(tpu)。聚氨酯是化学和/或物理交联的缩聚物,其典型地由多元醇和异氰酸酯形成并含有软链段和硬链段。软链段例如由聚酯、聚醚、聚碳酸酯(在本发明的上下文中各自优选地在性质上为脂族的)和多异氰酸酯硬链段组成。根据单独组分的性质和使用比率,可在本发明的上下文中有利地使用的材料是可获得的。例如在ep 894 841 b1和ep 1 308 492 b1中提出配制师为此可用的原料。
[0197]
也可用作基体聚合物(b)的热塑性弹性体的是基于聚烯烃的热塑性弹性体、聚醚酯弹性体、聚酰胺如聚酯酰胺、聚醚酯酰胺、聚碳酸酯酯酰胺和聚醚-嵌段-酰胺。
[0198]
如果使用基体聚合物(b),则选择如下的那些:其在引发固化过程之前基本上不参与与环氧官能团的反应或特别地完全不参与与环氧官能团的反应或者不引发或不催化环氧官能团的反应或者与环氧官能团的反应已经以一些其它的方式得到抑制。
[0199]
增粘树脂(c)
[0200]
本发明的环氧官能化(共)聚合物任选地与至少一种类型的增粘树脂、有利地与可与环氧官能化(共)聚合物和/或基体聚合物(b)相容的那些组合。在总胶粘剂组合物中的比例为至多40重量%、优选地至多25重量%,且可甚至为0重量%。
[0201]
当该增粘树脂具有大于25℃、特别地大于80℃的增粘树脂软化温度(astm e28)时则为有利的。
[0202]
胶粘剂组合物中使用的增粘树脂(c)可为例如基于松香和松香衍生物的部分或完全氢化或歧化的树脂、茚-香豆酮树脂、萜烯-酚醛树脂、酚醛树脂、二环戊二烯的氢化的聚
合物、基于c5、c5/c9或c9单体流的部分、选择性或完全氢化的烃树脂、基于α-蒎烯和/或β-蒎烯和/或δ-柠檬烯的多(聚)萜烯树脂、优选地纯c8和c9芳族化合物的氢化的聚合物。上述增粘树脂可单独地或以混合物使用。
[0203]
为了确保高的老化和uv稳定性,优选具有至少90%、优选地至少95%的氢化水平的氢化树脂。
[0204]
另外优选的(特别地与非极性成膜剂组合)是具有高于30℃的dacp(双丙酮醇浊点)值和大于50℃的mmap(混合甲基环己烷-苯胺点)值、特别地具有高于37℃的dacp值和大于60℃的mmap值的非极性树脂。dacp值和mmap值各自表示在特定溶剂混合物中的溶解度。对于dacp值和mmap值的定义和测定,提及c.donker,pstc annual technical proceedings,第149-164页,2001年5月。关于mmap,也可参考astm c611。
[0205]
如果使用增粘树脂(c),则选择如下的那些:其在引发固化过程之前基本上不参与与环氧官能团的反应或特别地完全不参与与环氧官能团的反应或者不引发或不催化环氧官能团的反应或者与环氧官能团的反应已经以一些其它的方式得到抑制。
[0206]
低粘度反应性树脂(d)
[0207]
任选地但是有利地,在与本发明的环氧官能化(共)聚合物组合的情况下,可使用低分子量的反应性树脂。它们以至多50重量%、优选地至多25重量%、非常优选地至多10重量%的在总配制物中的比例使用。这些低粘度反应性树脂特别地为环醚,即具有至少一个环氧乙烷基团的化合物,或氧杂环丁烷。它们在性质上可为芳族的或特别地脂族或脂环族的。可用的反应性树脂可为单官能的、双官能的、三官能的或四官能的或具有更高官能度直至多官能度,其中官能度涉及环醚基团。
[0208]
不希望施加限制,实例为:3,4-环氧基环己基甲基-3
‘
,4
‘‑
环氧基环己烷羧酸酯(eec)及衍生物、二环戊二烯二氧化物及衍生物、3-乙基-3-氧杂环丁烷甲醇及衍生物、四氢邻苯二甲酸二缩水甘油酯及衍生物、六氢邻苯二甲酸二缩水甘油酯及衍生物、乙烷1,2-二缩水甘油醚及衍生物、丙烷1,3-二缩水甘油醚及衍生物、丁烷-1,4-二醇二缩水甘油醚及衍生物、高级烷烃1,n-二缩水甘油醚及衍生物、双[(3,4-环氧基环己基)甲基]己二酸酯及衍生物、乙烯基环己基二氧化物及衍生物、环己烷-1,4-二甲醇双(3,4-环氧基环己烷羧酸酯)及衍生物、4,5-环氧基四氢邻苯二甲酸二缩水甘油酯及衍生物、双[1-乙基(3-氧杂环丁烷基)甲基]醚及衍生物、季戊四醇四缩水甘油醚及衍生物、双酚a二缩水甘油醚(dgeba)、氢化双酚a二缩水甘油醚、双酚f二缩水甘油醚、氢化双酚f二缩水甘油醚、环氧基酚-酚醛清漆、氢化环氧基酚-酚醛清漆、环氧基甲酚-酚醛清漆、氢化环氧基甲酚-酚醛清漆、2-(7-氧杂二环螺(1,3-二烷-5,3'-(7-氧杂二环[4.1.0]-庚烷))、1,4-双((2,3-环氧基丙氧基)甲基)环己烷。在此也优选脂(环)族环氧化物。
[0209]
根据wo 2013/156509 a2的化合物同样可在本发明的上下文中用作反应性树脂。
[0210]
反应性树脂可以其单体形式或二聚体形式、三聚体形式等直至其低聚体形式使用,尤其是如果重均分子量未达到5000g/mol。由于这些反应性树脂典型地具有低粘度,它们存在如下的风险:其在胶粘剂配制物中的过高比例将导致过高的挤出倾向性。因此,优选最低的在总配制物中的比例(至多25重量%、优选地至多10重量%)。仅当环氧官能化(共)聚合物的比例为至少50重量%且(共聚)单体(a)和任选地(b)在环氧官能化(共)聚合物中的比例总和为至少50重量%时,可使用最高达50重量%。
[0211]
反应性树脂彼此的混合物或者反应性树脂与其它共反应性化合物如醇(单官能或多官能的)或乙烯基醚(单官能或多官能的)的混合物同样是可能的。也可完全摒弃低分子量的反应性树脂。
[0212]
填料(e)
[0213]
如果与环氧官能化(共)聚合物组合使用填料颗粒,它们可优选地具有球形、棒状或片状结构。分离的颗粒(常常也称为一次颗粒)在此以与由多个一次颗粒形成的聚集体相同的方式同样为本发明所涵盖。这种体系常常呈现不规则碎片形(fraktale)上部结构。如果颗粒由微晶形成,则一次颗粒形式取决于晶格的类型。片形式的体系也可为层堆叠体的形式。如果使用填料,则典型地以最高达15重量%的程度使用它们。
[0214]
在本发明的有利实施方式中,胶粘剂配制物中的一种类型的填料基本上为单独的球形颗粒的形式。在该情况下,粒径具有小于500nm、优选地小于100nm、非常优选地小于25nm的值。在本发明的另一有利形式中,至少一种官能化类型的填料基本上以单独的片形式的颗粒的形式存在于胶粘剂组合物中。在该情况下,这样的片具有优选地小于10nm的值的层厚和优选地小于1000nm的最大直径。在本发明的另一有利形式中,至少一种类型的填料基本上以单独的棒形式的颗粒的形式存在于胶粘剂组合物中。在这种情况下,棒具有小于100nm的直径和小于15μm的长度。棒也可为弯曲的形式和/或柔性的。此外,在本发明的上下文中可为有利的是:胶粘剂组合物中的至少一种类型的填料为一次颗粒聚集体的形式。这些聚集体具有小于1000nm、优选地小于250nm的回转半径(与从聚合物已知的术语“回转半径”类似地理解)。在本发明的上下文中,特别优选使用具有小于250nm、优选地小于100nm、非常优选地小于50nm的在至少一个方向上的空间范围(尺寸)的那些填料颗粒。在本发明的上下文中,还可使用前述类型的填料的组合。
[0215]
对于填料,根据本发明有利的典型的和另外的化合物类别是无机氧化物、特别地金属氧化物和/或半金属氧化物、碱土金属盐和基于硅酸盐的矿物、特别地粘土矿物和粘土。根据本发明可使用的无定形或结晶金属氧化物包括例如二氧化硅、氧化铝、二氧化钛、二氧化锆和氧化锌。本领域技术人员熟悉可同样根据本发明使用的其它体系。碱土金属盐包括例如镁、钙、锶和钡的碳酸盐、硫酸盐、氢氧化物、磷酸盐和磷酸氢盐。根据本发明可使用的粘土矿物和粘土特别包括硅酸盐体系如蛇纹石、高岭土、滑石、叶蜡石、蒙皂石,如特别地蒙脱石、蛭石、伊利石、云母、脆性云母、亚氯酸盐、海泡石和坡缕石。此外,根据本发明,可使用合成粘土矿物如锂蒙脱石及其相关体系如来自laporte的和氟代锂蒙脱石及其相关体系如来自co-op的
[0216]
填料颗粒在其表面上可为官能化的,并且可为疏水化的或亲水化的。特别有利的官能化是借助于可参与固化反应的含环氧基的化合物。
[0217]
填料不是强制性的;即使在不单独地或以任何组合添加这些的情况下胶粘剂组合物也起作用。而且在任选的填料当中,选择如下的那些:其在引发固化过程之前基本上不参与与环氧官能团的反应或特别地完全不参与与环氧官能团的反应或者不引发或不催化环氧官能团的反应,或者与环氧官能团的反应已经以一些其它方式得到抑制。
[0218]
粘附促进剂(f)
[0219]
与基于硅烷的共聚单体(d)(如果这些被使用的话)组合,或者替代地,与环氧官能化(共)聚合物组合使用的粘附促进剂可为未通过聚合引入本发明的环氧官能化(共)聚合
物中的另外的硅烷。
[0220]
不希望施加限制,在本发明的上下文中可使用的硅烷的实例是甲基三甲氧基硅烷、甲基三乙氧基硅烷、二甲基二甲氧基硅烷、二甲基二乙氧基硅烷、三甲基乙氧基硅烷、乙基三甲氧基硅烷、丙基三甲氧基硅烷、丙基三乙氧基硅烷、异丁基三甲氧基硅烷、异丁基三乙氧基硅烷、辛基三甲氧基硅烷、辛基三乙氧基硅烷、异辛基三甲氧基硅烷、异辛基三乙氧基硅烷、十六烷基三甲氧基硅烷、十六烷基三乙氧基硅烷、十八烷基甲基二甲氧基硅烷、苯基三甲氧基硅烷、苯基三乙氧基硅烷、环己基甲基二甲氧基硅烷、二环戊基二甲氧基硅烷。
[0221]
可根据本发明使用的甲硅烷基官能化低聚物或聚合物的一个实例是与三甲氧基硅烷基团连接的聚乙二醇。
[0222]
可用的具有至少一个官能团的硅烷的另外的实例是乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、乙烯基三(2-甲氧基乙氧基)硅烷、乙烯基三异丙氧基硅烷、乙烯基二甲氧基甲基硅烷、乙烯基三乙酰氧基硅烷、3-缩水甘油氧基丙基三甲氧基硅烷、3-缩水甘油氧基丙基三乙氧基硅烷、2-(3,4-环氧基环己基)乙基三乙氧基硅烷、3-缩水甘油氧基丙基二乙氧基甲基硅烷、3-甲基丙烯酰氧基丙基三甲氧基硅烷、3-甲基丙烯酰氧基丙基三乙氧基硅烷、3-甲基丙烯酰氧基丙基三异丙氧基硅烷、3-甲基丙烯酰氧基丙基二甲氧基甲基硅烷、3-甲基丙烯酰氧基丙基二乙氧基甲基硅烷、3-氯丙基三甲氧基硅烷、3-氯丙基三乙氧基硅烷、3-脲丙基三甲氧基硅烷、3-脲丙基三乙氧基硅烷、2-羟基-4-(3-三乙氧基甲硅烷基丙氧基)二苯甲酮、4-(3'-氯二甲基甲硅烷基丙氧基)二苯甲酮。
[0223]
另外的添加剂(g)
[0224]
可作为掺混物/添加剂(g)添加至环氧官能化(共)聚合物或基于其的胶粘剂组合物的另外的任选成分为惯常的掺混物如老化稳定剂(抗臭氧剂、抗氧化剂、光稳定剂等)。
[0225]
可能的胶粘剂体系的添加剂包括如下:
[0226]
·
主抗氧化剂,例如空间位阻酚,
[0227]
·
辅抗氧化剂,例如亚磷酸酯(盐)或硫醚
[0228]
·
加工稳定剂,例如c-自由基清除剂
[0229]
·
光稳定剂,例如uv吸收剂或空间位阻胺
[0230]
·
加工助剂,如流变活性添加剂(例如增稠剂)
[0231]
·
润湿添加剂
[0232]
·
发泡剂如化学发泡剂和/或膨胀或可膨胀微球和/或中空珠如中空玻璃珠
[0233]
·
粘附促进剂
[0234]
·
增容剂
[0235]
·
着色剂/颜料
[0236]
掺混物或添加剂不是强制性的;本发明的胶粘剂组合物的一个优点是:即使在不单独地或以任何组合添加另外的添加剂的情况下,它也具有其有利的性质。然而,在特定情况下如下可为有利的和合乎期望的:通过添加添加剂来调节胶粘剂组合物的特定的其它性质。
[0237]
而且在任选的掺混物/添加剂当中,选择在引发固化反应之前基本上不参与与环氧官能团的反应或特别地完全不参与与环氧官能团的反应,或既不引发也不催化环氧官能团的反应的那些,或其中与环氧官能团的反应已经以一些其它方式得到抑制的那些。
[0238]
应用实例
[0239]
在其中可非常有利地使用本发明的环氧官能化(共)聚合物的应用的实例包括下面的实施方式。基于包括本发明的环氧官能化(共)聚合物的配制物的反应性胶粘剂体系特别适合以包含至少一个该胶粘剂体系的层的胶粘膜的形式使用。
[0240]
在这样的反应性胶带中,至少一个本发明的可固化反应性胶粘剂组合物层的层厚典型地在至少10μm和至多500μm之间、优选地在至少20μm和至多250μm之间。可用的层厚为30μm、50μm、75μm、100μm、125μm、150μm和200μm(各自在惯常的误差公差内)。
[0241]
反应性胶带特别地为双面胶粘剂产品,然而单面胶粘剂产品同样是可能的。在最简单的情况下,包括至少一个具有环氧官能化(共)聚合物的可固化胶粘剂组合物层的这种胶粘剂产品以单层形式使用(使得可固化胶粘剂组合物层和反应性胶带是相同的)、施加至另外的可分离(临时)载体材料。可用的临时载体材料包括从现有技术已知的并且在一侧或两侧配备有剥离层的所有剥离膜和剥离纸。纸也可单面或双面地涂覆有聚乙烯或聚丙烯。还可使用两个可再分离的载体材料的层,使得即使当产品不是卷绕形式时,胶粘膜的顶侧和底侧也被覆盖。临时载体材料不是经粘合的复合物的一部分。在粘合基底之前将其从反应性胶带除去。
[0242]
包括至少一个本发明的可固化胶粘剂组合物层的反应性胶带可另外地包含甚至在粘合之后也为复合物的一部分的另外的载体材料(永久性载体)。为此,同样地膜和纸以及铺设稀松布、织造和针织织物是可能的选择。这些载体材料的表面可各自独立地经过化学预处理(底漆、等离子体)和/或物理预处理(电晕、火焰、等离子体),使得可实现特别好的可固化胶粘膜层对载体材料的锚定。为了改善对永久载体材料的锚定,胶粘剂组合物也可被物理预处理(电晕、火焰、等离子体)。非织造织物是优选的。非织造织物的基重优选地在4和100g/m2之间、更优选地在10和70g/m2之间。这些非织造织物的厚度优选地在20和100μm之间、格外优选地在30和60μm之间。
[0243]
具有永久性载体的反应性胶带可在顶侧和/或底侧上承载不同厚度的可固化胶粘剂组合物层和/或优选地不同类型的可固化胶粘剂组合物层。如果使用不同的可固化胶粘剂组合物层,则两者都特别地包含至少一种本发明的(共)聚合物。
[0244]
包括至少一个具有本发明的(共)聚合物的可固化胶粘剂组合物层的反应性胶带也可以双层或多层和无永久性载体的形式使用。优选地最上层和非常优选地还有最下层是包括本发明(共)聚合物的可固化胶粘剂组合物层,其中这些在厚度和/或类型方面可不同。如果使用不同的可固化胶粘膜层,则两者都特别满足关于本发明的可固化胶粘剂组合物所做出的说明。在具有或不具有永久性载体的多层反应性胶带的情况下,原则上可能的其它实施方式是在顶侧上具有本发明的可固化胶粘剂组合物并且在底侧上具有另外的胶粘剂(例如压敏胶粘剂或热熔性胶粘剂)层的那些。
[0245]
多层的且含永久性载体的反应性胶带可具有30μm-1000μm、优选地75μm-300μm的厚度。
[0246]
反应性胶带可以幅材形式修整为卷材、片材或以模切件形式修整,并且可原样用于构造复合物。反应性胶带优选地在室温下不是压敏胶粘性的,因为即使在没有临时载体(例如模切件)的情况下材料也因而可非常有利地修整并且提供用于进一步的加工操作。然而,压敏胶粘性的设计也是可想到的和有利的。
[0247]
也是本发明的一部分的是包括如下的复合物:第一粘合基底、第二粘合基底和布置在它们之间的本发明的反应性胶带,该反应性胶带包括至少一个本发明的环氧官能化(共)聚合物层。在最终的复合物中,反应性胶带为固化状态。举例而言,将提及使用本发明的反应性胶带制备这样的复合物的典型手段。
[0248]
在最简单的情况下,将不具有临时载体的反应性胶带的模切件手动地例如借助于镊子定位在第一部件上或待组装部件之间。在另外的实施方式中,将反应性胶带的模切件在被定位在第一部件上之后用热源处理,由此增加模切件对第一部件的粘附性。这在层压温度下完成。在最简单的情况下,使用的热源可为ir源、扁铁或热板。对于该过程,有利的是,模切件仍配备有临时载体材料以防止胶粘膜对工具或热源的粘附。
[0249]
在另一有利的设计中,第一部件定位在反应性胶带的模切件上。在开放侧上进行定位。在背面上仍有临时载体材料。随后,热源将热通过第一部件引入到反应性胶带中。这在层压温度下进行。由此使胶粘膜呈粘性,并且与粘附至临时载体的情况相比,其更强力地粘附至第一部件。通过第一部件将其加热。
[0250]
在优选的形式中,对于热的引入,使用加热压机。加热压机的冲杆在此例如由铝、黄铜或青铜制成,并且其形状通常适应于部件的轮廓或模切件的尺寸。为了确保模切件在第一部件上的精确定位,通常,使用匹配于待粘合部件的轮廓的模具,由此防止滑动。在模具中的引导销(钉)和反应性胶带的临时载体材料中的相应引导孔可确保模切件和第一部件之间的精确定位。其它定位手段是可想到的。在热活化之后,从模具移出具有层压于其上的胶粘膜的第一部件。整个操作也可转换为自动操作。
[0251]
因此,用于制造本发明的复合物的方法还涉及包括如下步骤的子流程:
[0252]
a)将第一部件(基底)固定在成型部件(支架)上;
[0253]
b)使用包括至少一个本发明的可固化胶粘剂组合物层的反应性胶带将待粘合的第二部件(基底)放置在第二部件(基底)上;
[0254]
c)施加压力和温度,特别地借助于热压冲杆;
[0255]
d)从成型部件(支架)移除粘合的复合物,
[0256]
其中可选地也可在步骤c)和步骤d)之间进行再冷却。在步骤c)中,施加压力和温度。该温度为活化温度。这借助于由具有良好导热性的材料组成的加热冲杆进行。有利的材料为例如铜、黄铜、青铜或铝。但也可使用其它金属或合金。另外,热压冲杆应优选地呈现一个部件的顶侧的形状。该形状继而可在性质上为2维的或3维的。有利地经由气压缸(pneumatic cylinder)施加压力。然而,施加不必一定要经由空气压力进行。也可能的是例如水压设备或机电驱动器(例如,经由锭子)。另外,如下可为有利的:重复地引入压力和温度以例如通过串联连接或旋转原理增加过程吞吐量(工艺产量)。热压冲杆在该情况下无需全部用相同的温度和/或相同的压力进行操作。另外,也可选择不同的冲杆接触时间。对于活化,代替活化温度或与活化温度或另外的温度组合,也可使用(uv)辐射。
[0257]
对于基于所述胶粘剂体系的胶粘剂产品,多种用途是可想到的,其中在此可仅提及出于实例目的的选择,并且其不应视为限制:可热固化反应性胶带(例如用于高强度部件粘合)、可uv固化的反应性胶带(例如用于高强度部件粘合)、可热固化密封胶带、可uv固化的密封胶带。由于胶粘剂体系是基于聚(甲基)丙烯酸酯的,故而胶粘剂体系可以高的光学品质制造并且相应地用于其中这样的需求必须被满足的粘合任务。
[0258]
实验
[0259]
测试方法
[0260]
测试a
–
挤出(渗出)
[0261]
渗出测试使得能够得到关于胶粘剂组合物的挤出特性的结论(也参见图1)。对于程序也参见图1a和1b。为此,使用待检查的胶粘膜(3)将具有直径21mm的聚碳酸酯的圆形基底(1)粘合至阳极氧化铝的第二基底(2)。第二基底具有拥有直径9mm的圆孔;第一基底以居中的方式施加,其中胶粘剂产品在该孔上方。胶粘剂产品(3)(以转移胶带试样的形式)同样具有21mm的直径和100μm的厚度,并且相应地按尺寸切割或模切。
[0262]
检查由基底(1)(聚碳酸酯;macrolon 099)和基底(2)(阳极氧化铝;e6ev1)组成的复合物。基底(1)具有1.0mm的厚度以及基底(2)具有3.0mm的厚度。
[0263]
将胶粘剂产品/胶粘膜在70℃下预层压到基底(1),然后将复合物(基底(1)和胶粘剂产品(3))预层压到基底(2)上。预层压过程中的热作用(70℃)总时间不得超过30秒。随后,在压力和温度下作用压制整个复合物。以结果记录温度、压力和压制时间。选择的压制条件是180℃,12秒,10巴。
[0264]
在压制之后,测定胶粘膜的挤出特性。在彼此成直角的四个位置(“北”、“东”、“南”和“西”)处,在各情况下在径向方向上,基于圆形聚碳酸酯基底,用具有1/10mm刻度的精密刻度放大镜测量从聚碳酸酯基底边缘到相应侧上的挤出的组合物(oz)的最外边缘的最大距离a。结果为来自四次单独测量的平均值。
[0265]
对于许多应用,需要最小的挤出倾向性。根据本测试方法学的渗出为至多1.5mm、优选地0mm。
[0266]
测试b
–
分子量gpc
[0267]
使用经澄清过滤的100μl样品(样品浓度1.5g/l)测定分子量。所用的洗脱剂是具有0.1体积%三氟乙酸的四氢呋喃,并且内标为200ppm(m/v)甲苯。测量在25℃下进行。
[0268]
使用的预柱是如下的柱:pss-sdv型,10μm,id 8.0mm x 50mm(在此和在下文中的值按如下的顺序:类型,颗粒尺寸,内径
×
长度)。使用如下的柱完成分离:pss
–
sdv型,10μm线性的柱,id 8.0mm x 300mm(来自的polymer standards service的柱和检测器;借助于pss-seccurity 1260 rid检测器的检测)。流速为0.5ml/分钟。在柱的分离区域中用聚苯乙烯标准物进行校正,并利用已知的mark-houwink系数a和k将其普遍转换为聚甲基丙烯酸甲酯校正。
[0269]
测试c
–
玻璃化转变温度(dsc)
[0270]
使用netzsch的dsc 204f1借助于动态差示量热法(dsc:差示扫描量热法)测定玻璃化转变温度(tg)。将样品称重到增强的铝坩埚(手动穿孔的盖子)中。温度程序运行两次加热升温,首先用液氮从25℃冷却到-100℃并且以10k/分钟加热直至180℃。玻璃化转变被认为是温谱图中的台阶。玻璃化转变温度如下评价(在这方面,参见图2)。在各情况下,在台阶的1之前和2之后将切线施加至温谱图的基线。在台阶的区域中,将最佳拟合线3以这样的方式平行于纵坐标安置,使得两条切线相交,具体地例如以形成两个等面积的区域4和5(在相应切线、最佳拟合线、和测量图之间)。如此定位的最佳拟合线与测量图的交点给出玻璃化转变温度。
[0271]
随后,将样品冷却回到-100℃并以10k/分钟加热直至250℃。评价第一和第二加热
升温。由此在第一加热曲线中确定的玻璃化转变温度对应于未交联的聚合物的玻璃化转变温度。由第二加热曲线得到的所确定的玻璃化转变温度对应于通过测量的热应力而交联的聚合物、或通过热交联剂/引发剂(在其存在于聚合物或配制物中的情况下)的活化而交联的聚合物或配制物的玻璃化转变温度。对于非反应性体系,也可以此方式确定玻璃化转变温度。于是将第二加热曲线中的台阶(梯级)评价为结果。
[0272]
测试d
–
活化温度(dsc)
[0273]
经由差示扫描量热法(dsc)测定可阳离子固化的反应性树脂的热固化所需的活化温度。在具有穿孔盖子的al坩埚和氮气气氛中分析试样。为了用样品实现坩埚底座的良好覆盖,首先将仪器中的试样加热直至40℃并冷却回到25℃。实际测量在25℃开始;加热曲线以10k/分钟的加热速率运行。评价第一加热曲线。热引发的固化反应的开始由测量设备通过释放的相关反应焓记录并且表示为温谱图中的放热信号(峰)。使用的活化温度是在该信号中的如下温度:在该温度下测量曲线开始偏离基线(温谱图的一阶导数可用作寻找该点的辅助;反应的起始可连接至温谱图中的点,在该点处起始区域中的峰的一阶导数与温谱图基线的一阶导数之间的差呈现0.01mw/(k分钟)的值;如果该图显示向上的放热信号,则该符号为正;如果它们显示向下的方向,则符号为负)。另外,记录对称重的试样量标准化的积分。
[0274]
测试e
–
耐推出性
[0275]
推出测试使得能够得到关于胶粘剂产品在胶粘剂层法线方向上的粘合强度的结论。为此,用待检查的胶粘膜将具有直径21mm的圆形基底(1)粘合至第二基底(1或2)。第二基底具有直径9mm的圆孔;第一基底以居中的方式施加,其中胶粘剂产品在该孔上方。胶粘剂产品同样具有21mm的直径,并且相应地按尺寸切割或模切。
[0276]
检查由基底(1)(聚碳酸酯;macrolon099)和基底(2)(阳极氧化铝;e6ev1)组成的复合物。基底(1)具有1.0mm的厚度以及基底(2)具有3.0mm的厚度。
[0277]
将胶粘剂产品/胶粘膜在70℃下预层压到基底(1)上,然后将复合物(基底(1)和胶粘剂产品)预层压到基底(2)上。预层压过程中的热作用(70℃)总时间不得超过30秒。随后,在压力和温度作用下压制整个复合物。以结果记录温度、压力和压制时间。选择的压制条件为180℃,12秒,10巴。
[0278]
借助于夹在拉伸测试机中的圆柱形冲杆(直径7mm),通过基底(2)中的孔压在复合物(基底(1)和胶粘剂产品)上,并由此对复合物中的胶粘粘合施加力。将基底(2)固定在拉伸测试机中,使得确保尽可能在所有侧上的平坦的铺设/固定,其中可通过冲杆自由地推出基底(1)。测试速度为10mm/秒。记录如下的力:在该力下粘合失效并且基底(1)从基底(2)分离。该力基于粘合面积(282mm2),从而结果是以n/mm2为单位的耐推出性。测试条件为23℃和50%相对空气湿度;在测试条件下压制48小时后储存试样。结果是来自三次单独测试的平均值并以n/mm2报告。在许多应用中,需要高粘合强度。根据本测试的良好粘合强度的指示是至少2n/mm2,且对于非常好的粘合强度为至少4.0n/mm2。
[0279]
实施例
[0280]
使用的原料:
[0281]
52来自dupont的2,2-偶氮双(2,4-二甲基戊腈)
[0282]
tta15来自tetrachem的甲基丙烯酸3,4-环氧基环己基甲酯
[0283]
cxc1614来自kingindustries的基于三氟甲磺酸的季铵盐的热活化剂
[0284]
530来自covestro的聚氨酯
[0285]
uvacure1500来自allnex的(3',4'-环氧基环己烷)甲基(3,4-环氧基-环己基)羧酸酯
[0286]
胶粘剂组合物和反应性胶带试样的制造:
[0287]
实施例a
[0288]
用100g甲基丙烯酸3,4-环氧基环己基甲酯和396g甲乙酮装填用于自由基聚合的常规类型的耐压2l聚合反应器。在搅拌的同时使氮气通过45分钟后,将反应器加热直至产物温度70℃并抽空至沸腾。随后,添加溶解在4.0g甲乙酮中的2.0g2,2-偶氮双(2,4-二甲基戊腈)。在蒸发冷却下在70℃的恒定产物温度下进行反应。在1小时的反应时间后,添加已经预加热至70℃并且已经将氮气通过其45分钟的100g甲基丙烯酸3,4-环氧基环己基甲酯,并且添加溶解在4.0g甲乙酮中的2.0g2,2-偶氮双(2,4-二甲基戊腈)。在2小时的反应时间后,添加已经预加热至70℃并且已经将氮气通过其45分钟的100g甲基丙烯酸3,4-环氧基环己基甲酯,并且添加溶解在4.0g甲乙酮中的2.0g2,2-偶氮双(2,4-二甲基戊腈)。在3小时的反应时间之后,添加已经预加热至70℃并且已经将氮气通过其45分钟的100g甲基丙烯酸3,4-环氧基环己基甲酯,并且添加溶解在4.0g甲乙酮中的2.0g2,2-偶氮双(2,4-二甲基戊腈)。在24小时反应时间后将反应终止并冷却至室温。
[0289]
所得聚合物的分子量为15900g/mol。
[0290]
未固化的聚合物的玻璃化转变温度为32℃,根据测试c由第一加热曲线测定。在dsc实验中在加热阶段期间通过自固化产生的材料在第二加热曲线中具有72℃的玻璃化转变温度。
[0291]
实施例b
[0292]
用400g甲基丙烯酸3,4-环氧基环己基甲酯、420g异丙醇和726g甲乙酮装填用于自由基聚合的常规类型的耐压2l聚合反应器。在搅拌的同时使氮气通过45分钟后,将反应器加热直至产物温度65℃并抽空至沸腾。随后,添加溶解在8.0g甲乙酮中的4.0g2,2-偶氮双(2,4-二甲基戊腈)。在蒸发冷却下在65℃的恒定产物温度下进行反应。在7小时的反应时间后,添加溶解在8.0g甲乙酮中的4.0g2,2-偶氮双(2,4-二甲基戊腈)。在24小时反应时间后将反应终止并冷却至室温。
[0293]
所得聚合物的分子量为25900g/mol。
[0294]
未固化的聚合物的玻璃化转变温度为34℃,根据测试c由第一加热曲线测定。在dsc实验中在加热阶段期间通过自固化产生的材料在第二加热曲线中具有68℃的玻璃化转变温度。
[0295]
实施例c
[0296]
用400g甲基丙烯酸3,4-环氧基环己基甲酯、420g异丙醇和150g甲乙酮装填用于自由基聚合的常规类型的耐压2l聚合反应器。在搅拌的同时使氮气通过45分钟后,将反应器加热直至产物温度65℃并抽空至沸腾。随后,添加溶解在30.4g异丙醇中的1.6g2,2-偶氮双(2,4-二甲基戊腈)。在蒸发冷却下在65℃的恒定产物温度下进行反应。在7小时的反应时间后,添加溶解在30.4g异丙醇中的1.6g2,2-偶氮双(2,4-二甲基戊腈)。在14小时的反应
时间后,用100g甲乙酮稀释混合物。在24小时反应时间后将反应终止并冷却至室温。
[0297]
所得聚合物的分子量为30 600g/mol。
[0298]
未固化的聚合物的玻璃化转变温度为38℃,根据测试c由第一加热曲线测定。在dsc实验中在加热阶段期间通过自固化产生的材料在第二加热曲线中具有70℃的玻璃化转变温度。
[0299]
实施例e
[0300]
为了制造反应性胶带试样(实施例e1和e2,对比例v1),分别将所需的所有配制物成分溶解在溶剂中并且用分散盘将任何不溶性成分如无机填料悬浮并作为溶液或悬浮体涂覆。溶液中的溶剂含量为80重量%。使用的溶剂是甲乙酮。在硅化剥离纸上进行涂覆。将经涂覆和干燥的试样在50℃下干燥30分钟。干燥后,涂层的胶粘剂层厚度为100μm(在惯常的误差公差内)。24小时后,加工反应性胶带试样以得到测试试样,然后在另外的48小时后测量。测试试样的细节可在相应的测试方法中找到。
[0301]
实施例e1-可热固化的反应性胶带
[0302]
将9.7重量%来自实施例a的聚合物与90重量%作为成膜剂(b)的desmomelt 530和0.3重量%作为固化剂(a)的k-pure cxc 1614掺混。对于所得的反应性胶带,检测粘合强度(根据测试e的耐推出性)和压制步骤中的挤出倾向性(测试a)。耐推出性为5.5n/mm2且挤出倾向性为0mm。
[0303]
实施例e2-可热固化的反应性胶带
[0304]
将49.7重量%来自实施例a的聚合物与50重量%作为成膜剂(b)的desmomelt 530和0.3重量%作为固化剂(a)的k-pure cxc 1614掺混。对于所得的反应性胶带,检测粘合强度(根据测试e的耐推出性)和压制步骤中的挤出倾向性(测试a)。耐推出性为2.2n/mm2且挤出倾向性为1.4mm。
[0305]
对比例v1-可热固化的反应性胶带
[0306]
将9.7重量%uvacure 1500(低分子量的脂环族二环氧化物)与90重量%作为成膜剂(b)的desmomelt 530和0.3重量%作为固化剂(a)的k-pure cxc1614掺混。配制物不含任何本发明的(共)聚合物。对于所得的反应性胶带,检测粘合强度(根据测试e的耐推出性)和压制步骤中的挤出倾向性(测试a)。耐推出性为2.5n/mm2且挤出倾向性》1.5mm。
[0307]
从实施例和对比例明晰,本发明的环氧官能化(共)聚合物可有利地用于可固化胶粘剂组合物中。根据所述任务,以些(共)聚合物实现了粘合强度。挤出倾向性在有利的范围内。如果使用根据现有技术的低分子量的反应性树脂(uvacure 1500)代替本发明的环氧官能化(共)聚合物,则不满足对低挤出倾向性的要求。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1