地屈孕酮及其中间体化合物的制备方法与流程
地屈孕酮及其中间体化合物的制备方法
1.本技术是申请日为2021年9月8日、申请号为202111049973.0、发明名称为“中间体化合物及其制备方法和应用”的中国专利申请的分案申请。
技术领域
2.本技术涉及化学制药领域,尤其涉及地屈孕酮及其中间体化合物的制备方法。
背景技术:
3.地屈孕酮(dydrogesterone),又名去氢孕酮,化学名为9β,10α-孕甾-4,6-二烯-3,20-二酮,cas号:152-62-5,化学式如下:
[0004][0005]
地屈孕酮以孕甾烷为母核,孕甾烷具有如下的骨架结构,具有abcd四个环(从左到右,四个环依次定义为a、b、c和d),碳的标号(1-21)如下,在下文中记为c-1位、c-2位等。
[0006][0007]
地屈孕酮既广泛用于保胎及预防流产,还广泛用于治疗内源性孕酮不足引起的各种疾病,如:痛经、子宫内膜异位症、继发性闭经、月经周期不规则、功能失调性子宫出血、经前期综合征、孕激素缺乏所致先兆性流产或习惯性流产、黄体不足所致不孕症等。
[0008]
目前一些合成路线是从麦角甾醇出发,经过光化学合成10α构型中间体,然后经过沃氏氧化、双键转位、臭氧氧化、烯胺化、最后氧化获得地屈孕酮。但是光转化过程转化率低、分离困难,同时此过程需要用到臭氧氧化,存在安全风险,且副产物较多。另一些合成路线是通过光甾-4,7,22-三烯-3-酮为原料经4步反应来合成,由于每步收率低、起始原料难得到等缺点,基本上不具备工业化生产的可能性。还有一些合成路线以反式孕酮为原料,以四氯苯醌为氧化剂来合成,虽然该合成路线短,但所用的原料反式孕酮在天然产物中并不存在,需要通过合成才能得到,且目前合成很困难,并无工业化产品,因此目前也不具备工业化生产的可能。
技术实现要素:
[0009]
本技术的目的是提供一种可工业化合成地屈孕酮的生产工艺,原料易得、总产率高,容易放大至工业化生产。
[0010]
本技术的一方面提供一种中间体化合物,具有如下结构式:
[0011][0012]
其中,r1选自卤素或or3;
[0013]
r2选自=o、-or6或被保护的羰基;
[0014]
r3选自h、其中虚线表示与o连接位置;
[0015]
r4选自取代或未被取代的c1~c6直链或支链烷基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的苯基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的萘基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的吡啶基;
[0016]
r5选自取代或未被取代的c1~c6直链或支链烷基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的苯基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的萘基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的吡啶基;
[0017]
r6选自h或羟基保护基团;
[0018]
在结构式i中,表示化学键为单键或双键,且当某一为双键时,与其相邻的为单键。
[0019]
在本技术的一些实施例中,r4选自c1~c3直链或支链烷基,或者被c1~c3烷基取代或未被取代的苯基;和/或,r5选自c1~c3直链或支链烷基,或者被c1~c3烷基取代或未被取代的苯基,或者被c1~c3烷基取代或未被取代的萘基,或者被c1~c3烷基取代或未被取代的吡啶基;和/或,所述羟基保护基团选自-c(=o)r7、c1~c8烷基或c1~c8硅烷基;和/或,r7选自取代或未被取代的c1~c6直链或支链烷基;和/或,所述被保护的羰基选自缩酮;和/或,所述卤素或所述卤原子选自cl、br或i。
[0020]
在本技术的一些实施例中,r3选自h或如下基团:
[0021][0022]
在本技术的一些实施例中,所述中间体化合物包括如下结构式:
[0023][0024]
其中,r2选自-or6或被保护的羰基,r6’
选自c1~c8烷基。
[0025]
在本技术的一些实施例中,所述中间体化合物包括如下结构式:
[0026][0027][0028]
本技术的另一方面还提供一种制备中间体化合物的方法,所述中间体化合物为上述任一项所述的中间体化合物;所述方法包括:将结构式iia化合物进行光化学转化使c-10
位的甲基由β构型翻转为α构型得到结构式ⅱ化合物的步骤;化合物的步骤;
[0029]
在本技术的一些实施例中,结构式iia化合物中的r2选自-or6;所述结构式ⅱ化合物为结构式ⅱb化合物;
[0030][0031]
在本技术的一些实施例中,所述方法还包括:将所述结构式ⅱb化合物经c-3位羟基的氧化和c-5,6位双键移位得到结构式ⅲ化合物的步骤;
[0032][0033]
在本技术的一些实施例中,将所述结构式ⅱb化合物转化成所述结构式ⅲ化合物的方法包括:采用沃氏氧化反应使c-3位羟基氧化为酮基且5,6双键移位到4,5位;或者
[0034]
采用氧化试剂将所述结构式ⅱb化合物进行氧化处理,使所述结构式ⅱb化合物中的c-3位羟基氧化为酮基,所述氧化试剂的结构式如下:然后进行碱性处理,使5,6双键移位到4,5位,得到结构式ⅲ化合物。
[0035]
在本技术的一些实施例中,采用氧化试剂进行氧化处理,所述氧化试剂与结构式ⅱb化合物的摩尔比为(1.2~1.8):1。
[0036]
在本技术的一些实施例中,氧化处理时还添加碳酸氢盐和水,所述碳酸氢盐、所述水及所述结构式ⅱb化合物的摩尔比为(1.5~2.5):(0.8~1.2):1。
[0037]
在本技术的一些实施例中,采用有机胺进行碱性处理。
[0038]
在本技术的一些实施例中,所述方法还包括:将所述结构式ⅲ化合物双键移位得到结构式ⅳ化合物的步骤;
[0039][0040]
在本技术的一些实施例中,将结构式ⅲ化合物转化成结构式ⅳ化合物的方法包括:在质子酸条件下,使所述结构式ⅲ化合物的7,8位双键移位到6,7位,得到结构式ⅳ化合物;所述质子酸采用卤化氢的醇溶液的形式添加,所述醇包括乙醇、异丙醇、丁醇或乙二醇中的至少一种。
[0041]
在本技术的一些实施例中,将结构式ⅲ化合物转化成结构式ⅳ化合物的方法包括:将卤化氢的醇溶液加入含结构式ⅲ化合物的反应溶剂中,所述卤化氢的醇溶液的添加量为10v~15v,所述卤化氢的醇溶液中水的质量百分比小于0.2%,所述卤化氢的重量占所述卤化氢的醇溶液总重量的25wt%~40wt%。
[0042]
在本技术的一些实施例中,还向所述含结构式ⅲ化合物的反应溶剂中添加抗氧化剂,所述抗氧化剂的质量为所述结构式ⅲ化合物质量的0.8%~1.2%。
[0043]
在本技术的一些实施例中,所述中间体化合物为化合物e;所述方法还包括:将结构式ⅳ化合物经水解得到化合物e的步骤;
[0044]
结构式ⅳ化合物中的r1选自卤素或or3,r3选自
[0045]
在本技术还提供一种中间体化合物的应用,包括在上述任一项所述的中间体化合物的c-20位构造酮基以制备地屈孕酮。
[0046]
在本技术的一些实施例中,所述中间体化合物具有如下结构式或将所述中间体化合物转化为如下结构的中间体化合物:
[0047][0048]
所述构造酮基的方法包括:
[0049]
将所述中间体化合物的21位羟基氧化为醛基,得到化合物f;
[0050]
将所述化合物f的醛基进行烯胺化反应,然后将c-20位氧化为羰基,获得地屈孕酮;
[0051][0052]
本技术提供一种地屈孕酮中间体化合物的制备方法,所述制备方法包括:采用氧化试剂将如下结构式b的化合物进行氧化处理,使所述结构式b的化合物中的c-3位羟基氧化为酮基;然后进行碱性处理,使5,6双键移位到4,5位,以得到如下结构式c的中间体化合物
[0053][0054]
其中所述氧化试剂的结构式如下:
[0055][0056]
在本技术的一些实施例中,在进行所述氧化时还添加碳酸氢盐和水,所述碳酸氢盐、所述水及所述结构式b的化合物的摩尔比为(1.5~2.5):(0.8~1.2):1。
[0057]
在本技术的一些实施例中,所述碱性处理采用有机胺进行。
[0058]
在本技术的一些实施例中,所述地屈孕酮中间体化合物的制备方法包括:将结构式a的化合物进行光化学转化使c-10位的甲基由β构型翻转为α构型以得到结构式b的化合物的步骤:
[0059][0060]
在本技术的一些实施例中,所述光化学转化通过两步光化学转化反应来实现,其中所述两步光化学转化反应包括:使所述结构式a的化合物在第一波长的紫外光照射下开环,完成第一步光化学转化反应;使开环后的结构式a的化合物在第二波长的紫外光照射下闭环,完成第二步光化学转化反应。
[0061]
在本技术的一些实施例中,所述第一步光化学转化反应和所述第二步光化学转化
反应的反应溶剂为甲醇、乙醇、正己烷、石油醚、正庚烷、乙酸乙酯、四氢呋喃、乙二醇、异丙醇中的至少一种。
[0062]
本技术提供一种地屈孕酮的制备方法,所述制备方法包括:
[0063]
(1)在质子酸条件下,经反应将下式c所示的化合物中7,8位双键移位到6,7位,得到式d所示的化合物;
[0064]
(2)将所述式d所示的化合物的ots基团转化为酯基,然后在碱性条件下水解,得到式e所示的化合物;
[0065]
(3)将所述式e所示的化合物的21位羟基氧化为醛基,得到式f所示的化合物;
[0066]
(4)使所述式f所示的化合物中的醛基进行烯胺化反应,得到式g所示的化合物;
[0067]
(5)将所述式g所示的化合物的c-20位氧化为羰基,得到式h所示的地屈孕酮;
[0068][0069]
在本技术的一些实施例中,所述质子酸采用卤化氢的醇溶液的形式添加,所述醇包括乙醇、异丙醇、丁醇或乙二醇中的至少一种。
[0070]
在本技术的一些实施例中,所述卤化氢的醇溶液的添加量为10v~15v,所述卤化氢的醇溶液中水的质量百分比小于0.2%,所述卤化氢的重量占所述卤化氢的醇溶液总重量的25wt%~40wt%。
[0071]
本技术技术方案提供了一种本领域急需的制备地屈孕酮的新方法,该方法采用的起始原料可以由植物甾醇的发酵产物制得,来源广泛、绿色环保;本技术技术方案的中间体化合物只需进行ab环双键构建和侧链改造即可方便的合成地屈孕酮,总收率高、路线短,是一种可工业化的合成地屈孕酮的新工艺,因此解决了现有技术中原料来源、光转化过程转化率低、副产物较多、安全风险高、不易工业化生产的问题。
具体实施方式
[0072]
除非另有定义,本文所用所有技术和科学术语与本发明所属领域的普通技术人员通常理解的含义相同。若存在矛盾,则以本技术提供的定义为准。当本文中出现商品名时,意在指代其对应的商品或其活性成分。本文引用的所有专利、已经公开的专利申请和出版物均通过引用并入到本文中。
[0073]
术语“一个(种)或多个(种)”或者类似的表述“至少一个(种)”可以表示例如1、2、3、4、5、6、7、8、9、10个(种)或更多个(种)。
[0074]
本文所用的表述m-n指m至n的范围以及由其中的各个点值组成的亚范围以及各个点值。例如,表述“c1-c6”或“c1-6”涵盖1-6个碳原子的范围,并应理解为还涵盖其中的任意亚范围以及每个点值,例如c2-c5、c3-c4、c1-c2、c1-c3、c1-c4、c1-c5、c1-c6等,以及c1、c2、c3、c4、c5、c6等。
[0075]
术语“烷基”是指由碳原子和氢原子组成的直链或支链的饱和的脂肪烃基团,其通过单键与分子的其余部分连接。“烷基”可以具有1-8个碳原子,即“c1-8烷基”,例如c1-4烷基、c1-3烷基、c1-2烷基、c3烷基、c4烷基、c1-6烷基、c3-6烷基。烷基的非限制性实例包括但不限于甲基、乙基、丙基、丁基、戊基、己基、异丙基、异丁基、仲丁基、叔丁基、异戊基、2-甲基丁基、1-甲基丁基、1-乙基丙基、1,2-二甲基丙基、新戊基、1,1-二甲基丙基、4-甲基戊基、3-甲基戊基、2-甲基戊基、1-甲基戊基、2-乙基丁基、1-乙基丁基、3,3-二甲基丁基、2,2-二甲基丁基、1,1-二甲基丁基、2,3-二甲基丁基、1,3-二甲基丁基或1,2-二甲基丁基,或者它们的异构体。
[0076]
术语“硅烷基”,是指如上定义的烷基,其中至少一个c原子被si原子取代。硅烷基与分子的其他部分通过硅原子相连。“c1-8硅烷基”是指含有1-8个碳原子的硅烷基,其中的烷基部分可为直链、支链或环状结构。硅烷基包括但不仅限于,三甲基硅基(tms)、叔丁基二甲基硅基(tbs,或称tbdms)、二甲基异丙基硅基(ipdms)、二叔丁基甲基硅基等(dtbms)。
[0077]
本发明的化合物可以存在特定的几何或立体异构体形式。本发明设想所有的这类化合物,包括顺式和反式异构体、(-)-和(+)-对对映体、(r)-和(s)-对映体、非对映异构体、(d)-异构体、(l)-异构体,及其外消旋混合物和其他混合物,例如对映异构体或非对映体富集的混合物,所有这些混合物都属于本发明的范围之内。此类物质的纯化和分离可通过本领域已知的标准技术实现。
[0078]
下述发明详述旨在举例说明非限制性实施方案,使本领域其它技术人员更充分地理解本发明的技术方案、其原理及其实际应用,以便本领域其它技术人员可以以许多形式修改和实施本发明,使其可最佳地适应特定用途的要求。
[0079]
中间体化合物
[0080]
本技术实施例一方面提供一种中间体化合物,所述中间体化合物可以用于制备地屈孕酮,具有如下结构式:
[0081][0082]
在结构式i中,r1可以选自卤素或or3;r2选自=o、-or6或被保护的羰基;r3选自h、其中虚线表示与o连接位置;r4选自取代或未被取代的c1~c6直链或支链烷基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的苯基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的萘基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的吡啶基;r5选自取代或未被取代的c1~c6直链或支链烷基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的苯基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的萘基,或者被c1~c6烷基、羟基或卤原子取代或未被取代的吡啶基;r6选自h或羟基保护基团。
[0083]
在结构式i中,表示化学键为单键或双键,且当某一为双键时,与这个相邻的为单键。
[0084]
r4选自c1~c3直链或支链烷基,或者被c1~c3烷基取代或未被取代的苯基;和/或,r5选自c1~c3直链或支链烷基,或者被c1~c3烷基取代或未被取代的苯基,或者被c1~c3烷基取代或未被取代的萘基,或者被c1~c3烷基取代或未被取代的吡啶基;和/或,所述羟基保护基团选自-c(=o)r7、c1~c8烷基或c1~c8硅烷基;和/或,r7选自取代或未被取代的c1~c6直链或支链烷基;和/或,所述被保护的羰基选自缩酮;和/或,所述卤素或所述卤原子选自cl、br或i。
[0085]
在一些实施例中,r3选自h或如下基团:
[0086][0087]
在一些实施例中,所述中间体化合物包括如下结构式:
[0088][0089]
其中,r2选自-or6或被保护的羰基,r6选自h或羟基保护基团,所述被保护的羰基选自缩酮,r6’
选自c1~c8烷基。
[0090]
作为示例,所述结构式i的部分具体结构式如下:
[0091][0092][0093]
其中的某些化合物的羟基或羰基可以被保护基团所保护。例如:
[0094]
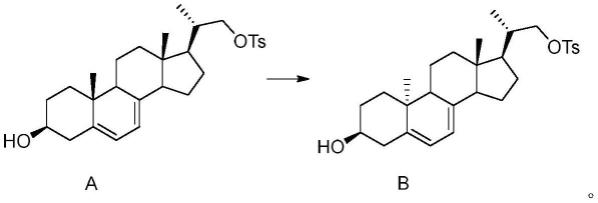
被保护后的结构式为
[0095]
被保护后的结构式为:
[0096][0097]
中间体化合物的制备方法
[0098]
(光化学转化的步骤)
[0099]
本技术实施例还提供一种制备前述任一项中间体化合物的方法。所述方法包括将结构式iia化合物进行光化学转化使c-10位的甲基由β构型翻转为α构型得到结构式ⅱ化合物的步骤;
[0100]
在一些实施例中,r2选自-or6。在一些实施例中,r6为h,所述结构式ⅱ化合物为结构式ⅱb化合物,所述将结构式ⅱa化合物转化成所述结构式ⅱb化合物通过两步光化学转化反应实现;
[0101][0102]
在一些实施例中,所述两步光化学转化反应可以包括:使所述结构式ⅱa化合物在第一波长的紫外光照射下开环,完成第一步光化学转化反应;使开环后的结构式ⅱa化合物在第二波长的紫外光照射下闭环,完成第二步光化学转化反应。
[0103]
可以采用植物甾醇为原料经分枝杆菌(mycobacterium)属的微生物发酵得到发酵产物(见下式),然后通过化学合成得到所述结构式ⅱa化合物。r2选自-or6时可以通过更短的步骤得到结构式ⅱa化合物。因此,本技术实施例的原料容易获取。
[0104][0105]
在一些实施例中,所述第一步光化学转化反应和所述第二步光化学转化反应的反应溶剂为甲醇、乙醇、正己烷、石油醚、正庚烷、乙酸乙酯、四氢呋喃、乙二醇、异丙醇中的至少一种,反应温度为-10℃至50℃;第一波长为270nm至290nm,所述第二波长为300nm至330nm。在一些实施例中,采用高压汞灯或led灯来提供紫外光。
[0106]
光化学转化会产生多个产物。r1为芳香基团时,例如r1为ots,该步骤的光化学转化反应具有更好的转化率和选择性(选择性达40%以上),可以取得更好的收率(收率可达25%),同时结构式ⅱb化合物更容易从产物中分离,后处理简便。
[0107]
结构式ⅱb化合物中的r1为-or3且r3选自时,可以先对c-3位羟基进行保护,再将r1转化为酯基,再脱保护,这样可以使酯化产物更易分离。
[0108]
(氧化和碱性条件双键移位的步骤)
[0109]
在一些实施例中,所述方法还包括:将所述结构式ⅱb化合物经c-3位羟基的氧化和c-5,6位双键移位得到结构式ⅲ化合物的步骤;
[0110][0111]
其中结构式ⅱb化合物的结构存在特殊性,如b环的共轭双键使结构性变差、c-10位α构型的甲基改变化合物溶解性、c-21位的基团的不同等,导致该步氧化反应对氧化体系要求较高。若采用一些常用的氧化体系,要么得不到目标产物,要么收率很低。例如采用斯文氧化可以得到结构式ⅲ化合物,但收率很低,杂质比较难控制且条件苛刻。
[0112]
可以采用沃氏氧化反应使c-3位羟基氧化为酮基且5,6双键移位到4,5位。沃氏氧化(试剂例如采用异丙醇铝/环己酮),其为高温反应,且存在必须高温才能除去的高沸点物质,虽然沃氏氧化可以同时进行双键移位,但结构式ⅲ化合物在高温或者强碱性条件下都不稳定,从而导致反应收率较低(摩尔收率约48%)。
[0113]
在一些实施例中,将所述结构式ⅱb化合物转化成所述结构式ⅲ化合物的方法可以包括:将所述结构式ⅱb化合物进行氧化处理,使所述结构式ⅱb化合物中的c-3位羟基氧化为酮基;然后进行碱性处理,使5,6双键移位到4,5位,得到结构式ⅲ化合物。
[0114]
本技术实施例采用的氧化试剂具有如下结构式(戴斯马丁试剂):
[0115]
能够在低温下(例如-5℃至25℃、或5℃至10℃)进行氧化反应。所述氧化试剂与结构式ⅱb化合物的摩尔比可以为(1.2~1.8):1。在一些实施例中,氧化处理时还添加水和碳酸氢盐(例如碳酸氢钠、碳酸氢钾),可以促进反应,增加转化率和收率,转化率高达92%,摩尔收率高达70%。所述碳酸氢盐、水与结构式ⅱb化合物的摩尔比为(1.5~2.5):(0.8~1.2):1。再采用有机碱,优选胺,例如三乙胺、吡啶等,在温和条件下进行碱性处理,可以获得较高收率的结构式ⅲ化合物,这种方法避免了高温、长时间浓缩、强碱的影响。
[0116]
(酸性条件双键移位的步骤)
[0117]
在一些实施例中,所述方法还包括:将所述结构式ⅲ化合物双键移位得到结构式ⅳ化合物的步骤;
[0118][0119]
在一些实施例中,将结构式ⅲ化合物转化成结构式ⅳ化合物的方法包括:在质子酸条件下,使所述结构式ⅲ化合物的7,8位双键移位到6,7位,得到结构式ⅳ化合物。
[0120]
由于采用盐酸、硫酸、高氯酸、冰醋酸、对甲苯磺酸、三氟乙酸等转化率都较低,在一些实施例中,所用质子酸可以是hcl,hbr等。
[0121]
由于化学结构的特殊性,该步反应如要获得高的转化率对水分含量、酸浓度、酸量也有比较苛刻的要求。在一些实施例中,将结构式ⅲ化合物转化成结构式ⅳ化合物的方法包括:将卤化氢的醇溶液加入含结构式ⅲ化合物的反应溶剂中,按1g结构式ⅲ化合物添加所述10ml~15ml的卤化氢的醇溶液的方式添加卤化氢的醇溶液,也即所述卤化氢的醇溶液的添加量为10v~15v。所述卤化氢的醇溶液中水的质量百分比小于0.2%,所述卤化氢的重量占所述卤化氢的醇溶液总重量的25wt%~40wt%。
[0122]
在所述卤化氢的醇溶液中,醇可以采用乙醇、异丙醇,丁醇,乙二醇等。采用乙醇转化率可高达89%,而同等条件下采用甲醇转化率只有约55%。
[0123]
在一些实施例中,该步骤还可以添加质量分数为0.8%~1.2%(基于结构式ⅲ化合物的质量)的抗氧化剂来抑制过氧化的杂质,以提高收率。抗氧化剂示例性的包括抗坏血酸钠、tbhq。
[0124]
此外,结构式ⅳ化合物的熔点较低,较难得到固体,且在高浓度酸下有一定程度变质成油,影响固体的性状。在本技术实施例中,为了避免此类现象发生,在后处理时采用乙醇作为溶剂边搅拌边梯度降温得到固体,粗品再用正庚烷打浆除油,可以获得较高的收率。
[0125]
(水解的步骤)
[0126]
在一些实施例中,所述中间体化合物为化合物e;所述方法还包括:将结构式ⅳ化合物经水解得到化合物e的步骤;
[0127][0128]
在一些实施例中,所述结构式ⅳ中的r1为-or3且r3为将所述结构式ⅳ化合物转化成所述结构式e化合物的方法包括:将所述结构式ⅳ化合物在碱性条件下水解,形成21位羟基结构,其中采用的碱性物质包括naoh、koh、醋酸钾、醋酸钠和苯甲酸钠中的至少一种。
[0129]
在一些实施例中,r1为-or3且r3为将所述结构式ⅳ化合物转化成所述结
构式e化合物的方法包括:采用dmf和koac使所述结构式ⅳ化合物中的r1转化为酯基;然后在碱性条件下水解,其中采用的碱性物质包括naoh、koh、醋酸钾、醋酸钠和苯甲酸钠中的至少一种。
[0130]
由中间体制备地屈孕酮的方法
[0131]
本技术实施例还提供一种制备地屈孕酮的方法,包括在本技术实施例的中间体化合物的c-20位构造酮基。
[0132]
在一些实施例中,所述中间体化合物具有如下结构式:
[0133][0134]
在所述中间体化合物的c-20位构造酮基的方法包括:
[0135]
将所述中间体化合物的21位羟基氧化为醛基,得到化合物f。氧化体系可以包括naclo、nabr和2,2,6,6-四甲基哌啶-1-氧自由基,且所述氧化体系的ph范围控制在8-9。
[0136][0137]
将所述化合物f的醛基进行烯胺化反应,得到化合物ga,其中r8、r9选自c1~c6烷基或r
8-n-r9构成5~7元氮杂环。例如使所述化合物f与1-(1-哌啶基)环己烯进行烯胺化反应,得到化合物g;
[0138][0139]
氧化所述化合物g,获得地屈孕酮。例如可以将所述化合物g在cu
+
催化下进行空气氧化,获得地屈孕酮。
[0140]
作为示例,本技术实施例制备地屈孕酮h的其中一条合成路线l1如下:
[0141][0142]
其中r2选自-or6。
[0143]
需要说明的是,上述合成路线l1中,每一个箭头代表一步或若干步。例如,r2为oh时可以直接经光转化得到iib,r2为其它基团时除光转化的步骤外还需一步或若干步(可在光转化之前或之后)得到iib。
[0144]
上述合成路线l1中不同化合物的r1在定义范围内可以相同或不同。为从形式上体现这一点,可以转换成下述路线:
[0145][0146]
其中r
10
、r
11
、r
12
、r
13
的定义与上述r1相同,在定义范围内各自独立的进行选择。在一些实施例中,r
10
选自or3;r
11
、r
12
、r
13
选自卤素或or3;r3选自在一些实施例中,r
10
为ots基团,光转化后再将ots转变为oac或br(此时ots、oac或br统一记为r
11
),然后再进行iib至iii的反应。在一些实施例中,r
10
、r
11
、r
12
为ots基团,在得到式iv化合物后将ots转变为oac(此时ots、oac统一记为r
13
),然后再水解。
[0147]
上述合成路线l1在制备地屈孕酮h时,采用的原料为结构式iia化合物。但是,在其他实施例中,也可不以结构式iia化合物作为制备地屈孕酮的初始原料,而以化合物e或化合物e之前的任一化合物(结构式iib化合物、结构式iii化合物、结构式iv化合物)作为初始原料。选择的初始原料不同,相应的合成路线可以基于合成路线l1进行相应的删减。作为示例,选择结构式iii化合物作为原料时,可以在合成路线l1的基础上删除部分路线iia
→
iib
→
iii。
[0148]
作为示例,使合成路线l1中,结构式iia的r1为ots,r2为oh,也即选择结构式iia化合物中的一个具体化合物a作为原料合成地屈孕酮h,得到合成路线l2:
[0149][0150]
实施例
[0151]
以所述合成路线l2为例,通过具体实施例来说明制备地屈孕酮的方法。
[0152]
实施例1:a
→b[0153]
在光化反应瓶里加40g化合物a和500ml四氢呋喃,5-10℃紫外灯光下先开环,波长范围为270nm~290nm,光照8小时,hplc监测原料:产物=70:20左右,再经紫外灯照射波长范围为300nm~330nm继续光照8h,hplc监测,原料:产物=55:35左右停止;有机相浓缩,甲醇置换至小体积,降温-20℃冷冻1~2小时,过滤,烘干得白色固体16g,主要为原料;母液浓缩,乙腈置换至小体积后出料,降温-20℃冷冻1~2小时,过滤,烘干得白色固体10g化合物b,一次收率约25%。
[0154]
经检测,1h nmr(400mhz,cdcl3)δ7.78(d,j=8.3hz,2h),7.34(d,j=8.1hz,2h),5.66-5.64(m,1h),5.43
–
5.41(m,1h),4.09(s,1h),3.96(dd,j=9.3,3.1hz,1h),3.83(dd,j=9.3,6.0hz,1h),2.49-2.45(m,5h),2.29-2.24(m,2h),1.67
–
1.47(m,15h),0.97(d,j=6.7hz,3h),0.72(s,3h),0.57(s,3h)。
[0155]
实施例2:b
→c[0156]
在1l的三口烧瓶里加戴斯马丁试剂(dmp)105g(0.28mol),搅拌中加水3.78g(0.21mol),碳酸氢钠35g(0.41mol)和500ml二氯甲烷(dcm),搅拌片刻,5℃~10℃加100g(0.21mol)化合物b,保温搅拌半小时,tlc显示原料反应生成化合物b1,降温-20℃冷冻1~2小时,过滤,滤饼用冷二氯甲烷适量淋洗至滤饼无产物,有机相依次用亚硫酸钠溶液和碳酸氢钠溶液,食盐水洗涤。
[0157]
有机相加100ml三乙胺,常温搅拌1~2小时,tlc显示化合物b1转化为化合物c,有机相依次用食盐水,1m稀盐酸,食盐水洗涤,有机相40℃浓缩,正庚烷置换至小体积,降温-20℃冷冻1~2小时,过滤,烘干得黄色固体70g,摩尔收率约70%。
[0158]
经检测,1hnmr为:1h nmr(400mhz,cdcl3)δ7.78(d,j=8.3hz,2h),7.34(d,j=
8.1hz,2h),5.80(s,1h),5.23
–
5.14(m,1h),3.96(dd,j=9.3,3.1hz,1h),3.83(dd,j=9.3,6.0hz,1h),3.01(ddd,j=25.3,22.6,11.6hz,2h),2.56
–
2.27(m,6h),2.21(dd,j=6.9,2.6hz,1h),2.00(dt,j=13.4,4.7hz,1h),1.93
–
1.11(m,13h),1.04(s,3h),0.97(d,j=6.7hz,3h),0.63
–
0.55(m,3h)。
[0159]
按上述反应步骤做如下表1的对比试验,其它条件与本实施例相同。
[0160]
表1实验条件和结果
[0161][0162]
由表1可知,戴斯马汀试剂用量1.3eq左右即可,增加用量转化率没有明显提升,反而增大后处理的难度,影响收率;溶剂改为氯仿,收率没有提高;添加碳酸氢钠,可以促进反应,增加转化率和收率,添加少量水,也可以促进反应,增加转化率和收率以及速率,其原理可能是预先形成更活泼的dmp氧化中间态。
[0163]
实施例3:c
→d[0164]
在1l的三口烧瓶里加840ml的无水乙醇,低温通入干燥的氯化氢气体,制备得无水乙醇/氯化氢溶液(水分要小于0.2%,含量约35%);在2l的三口烧瓶里,加70g(0.145mol)化合物c和700ml二氯甲烷以及0.7g特丁基对苯二酚(tbhq),溶清,氮气保护,0℃~10℃下,滴加840ml自制的无水乙醇/氯化氢溶液,控温反应1小时左右,tlc检测原料剩余小于3%,加纯净水淬灭反应,分液,有机相用碳酸氢钠溶液洗至ph=7~8,有机相50℃以下浓缩,乙醇置换,保留约500ml乙醇,边搅拌边梯℃降温,黄色固体析出,降温-20℃冷冻1~2小时,过滤,粗品再用正庚烷打浆,冷析过滤,烘干得米白色固体50g,摩尔收率约70%。
[0165]
经检测:1h nmr(400mhz,cdcl3)δ7.76(d,j=8.3hz,2h),7.33(d,j=8.1hz,2h),6.24
–
6.05(m,2h),5.65(s,1h),3.94(dd,j=9.3,3.0hz,1h),3.80(dd,j=9.2,6.0hz,1h),2.60
–
2.48(m,1h),2.44(s,3h),2.41
–
2.32(m,1h),2.27
–
2.14(m,1h),1.91
–
1.55(m,10h),1.40
–
1.29(m,1h),1.29
–
1.07(m,6h),0.98(d,j=6.7hz,3h),0.70(s,3h)。
[0166]
13
c nmr(101mhz,cdcl3)δ199.59(s),163.38(s),144.65(s),140.96(s),132.91(s),129.75(s),127.85(s),126.81(s),123.60(s),75.47(s),51.57(s),49.22(s),42.70(s),39.59(s),38.65(s),38.24(s),37.10(s),36.07(s),35.49(s),33.89(s),27.00(s),24.89(s),22.14(s),21.59(s),20.46(s),16.72(s),10.58(s)。
[0167]
质谱:c29h38o4s,482.9。
[0168]
按上述反应步骤做表2中的对比试验,其它条件与本实施例相同。
[0169]
表2实验条件和结果
[0170][0171][0172]
由表1可知,该反应对水分要求高,浓盐酸或者95%乙醇反应转化率低,需要干燥的氯化氢气体/无水乙醇体系反应才能大大提高转化率。该反应对酸的浓度有要求,酸含量较低(20%),转化率相对较低,酸含量在30-38%能到达到较好的效果。该反应对酸量有要求,10v~15v较为合适,太低转化率低,太高会导致产物降解,表现为后处理固体油性较重,难析出固体,收率降低。此外,反应跟酸的强度有关,浓硫酸,三氟乙酸以及三氟甲磺酸转化率也有差异。有趣的是无水甲醇做溶液,转化率也较低,四氢呋喃和异丙醇做溶剂,转化率也低于无水乙醇。反应加抗氧化剂,可以抑制过氧化的杂质,提高收率。
[0173]
实施例4:d
→
d1
[0174]
在500ml的三口烧瓶里50g(0.10mol)化合物d和200ml dmf,溶清,加50g(0.51mol)醋酸钾,100℃反应2小时,tlc检测原料反应完全,缓慢倒入1l水中,析出固体,搅拌1小时,过滤,烘干得黄色固体36g,摩尔收率约95%。
[0175]
经检测:1h nmr(400mhz,cdcl3)δ6.15(dt,j=19.5,7.4hz,2h),5.64(s,1h),4.07(dd,j=10.7,3.5hz,1h),3.76(dd,j=10.7,7.3hz,1h),2.59
–
2.44(m,1h),2.40(ddd,j=11.5,7.4,4.9hz,2h),2.24(ddd,j=13.1,5.2,1.9hz,1h),2.03(s,3h),1.95
–
1.67(m,7h),1.67
–
1.52(m,2h),1.43
–
1.31(m,2h),1.29
–
1.13(m,5h),1.00(d,j=6.6hz,3h),0.75(s,3h).
[0176]
13
c nmr(101mhz,cdcl3)δ199.40(s),171.22(s),163.31(s),141.05(s),126.73
(s),123.59(s),69.30(s),52.66(s),49.32(s),42.80(s),39.68(s),38.73(s),38.45(s),37.13(s),35.61(d,j=13.6hz),33.90(s),27.21(s),25.01(s),22.14(s),20.91(s),20.53(s),16.99(s),10.66(s).
[0177]
质谱:c24h34o3,371.0。
[0178]
实施例5:d1
→e[0179]
在250ml的三口烧瓶里加36g(0.10mol)化合物d1和180ml甲醇,氮气保护,降温0℃~5℃,加7.2g(0.18mol)氢氧化钠固体,控温小于25℃,加完后自然回室温反应,反应0.5~1小时。tlc监测反应完全。加醋酸中和,再缓慢滴加水180ml,析出固体,冰浴搅拌1小时,过滤,烘干得黄色固体30g,摩尔收率约95%。
[0180]
经检测:1h nmr(400mhz,cdcl3)δ6.17(dt,j=23.1,7.5hz,2h),5.65(s,1h),3.64(dd,j=10.5,3.2hz,1h),3.39(dd,j=10.5,6.6hz,1h),2.69
–
2.46(m,1h),2.46
–
2.32(m,2h),2.25(ddd,j=13.1,5.2,1.9hz,1h),1.82(dddd,j=16.7,14.9,13.7,9.7hz,6h),1.69
–
1.50(m,4h),1.38(ddd,j=17.9,12.4,4.6hz,2h),1.28
–
1.15(m,5h),1.05(d,j=6.6hz,3h),0.76(s,3h).
[0181]
13
c nmr(101mhz,cdcl3)δ199.62(s),163.54(s),141.31(s),126.72(s),123.56(s),67.78(s),52.27(s),49.36(s),42.72(s),39.73(s),38.93
–
38.34(m),37.17(s),35.55(s),33.93(s),27.32(s),25.05(s),22.15(s),20.57(s),16.65(s),10.70(s).
[0182]
质谱:c22h32o2,329.0。
[0183]
实施例6:e
→f→g→h[0184]
(1)e
→f[0185]
在250ml的三口烧瓶里加30g(91.3mmol)化合物e和150ml二氯甲烷,搅拌下加1.5g(9.6mmol)tempo和溶解好的1.08g溴化钠(10.5mmol)和30ml 5%的碳酸氢钠水溶液,氮气保护,降温0℃~5℃,滴加次氯酸钠,控温小于15℃,反应0.5~1小时。tlc监测反应完全。硫代硫酸钠溶液淬灭,搅拌10分钟,分液,有机相用食盐水洗涤一次,有机相50℃以下浓缩,石油醚置换,保留3v~5v石油醚。降温到0℃冷析2小时,过滤,滤饼用冰石油醚淋洗,烘干得28g固体化合物f,摩尔收率约92%。
[0186]1h nmr(400mhz,cdcl3)δ9.58(d,j=3.1hz,1h),6.30
–
6.04(m,2h),5.67(s,1h),2.53(dd,j=14.2,5.4hz,1h),2.50
–
2.32(m,3h),2.26(ddd,j=13.2,5.3,2.1hz,1h),1.98
–
1.78(m,5h),1.73
–
1.37(m,6h),1.36
–
1.22(m,4h),1.13(t,j=6.1hz,3h),0.80(s,3h).
[0187]
13
c nmr(101mhz,cdcl3)δ208.83(s),199.28(s),162.87(s),140.34(s),126.99(s),123.80(s),63.29(s),49.78(s),44.15(s),39.59(s),38.50(s),37.64(s),37.10(s),35.50(s),33.87(s),31.40(s),25.07(s),22.49(s),22.21(s),20.47(s),11.98(s).
[0188]
质谱:c22h30o2,327.0。
[0189]
(2)f
→g→
h(地屈孕酮)
[0190]
在100ml的三口烧瓶里加28g(85.8mmol)化合物f和42ml无水乙腈,搅拌下加22g(122mmol)环己烯哌啶(含量约90%),氮气保护,40℃搅拌溶清,加冰醋酸,继续反应3-6小时,降温到-20℃,冷析2小时,过滤,滤饼用冰乙腈淋洗,抽干,固体35℃真空干燥箱烘,得28g化合物g。
[0191]
在100ml的三口烧瓶里加0.42g(4.2mmol)氯化亚铜和42ml dmf,氮气置换三次,加热至65℃,氮气保护保温搅拌1小时,降温至室温备用。在500ml的三口烧瓶里加28g(71.2mmol)化合物g和280ml二氯甲烷,降温至0-5℃,加入氯化亚铜溶液,通入已干燥的空气,保持气体流量1l/min,反应4~8小时,tlc检测原料剩余小于2%,延长时间无变化则可停止。加入10%硫酸溶液淬灭,分液,加1%硫酸溶液洗涤有机相,有机相加0.43g醋酸,搅拌5分钟,再加6%亚氯酸钠溶液,室温搅拌30min,tlc原料几乎消失。加硫代硫酸钠淬灭,分液,有机相依次用0.5%氢氧化钠,食盐水洗涤,有机相50℃以下浓缩,水置换出料得粗品。粗品加280ml丙酮加热溶清,浓缩到小体积,降温到-20℃,冷析2小时,过滤,滤饼用冰丙酮淋洗,抽干,45℃烘箱烘干。得20g固体化合物f,摩尔收率约74.6%。
[0192]
经检测:1h nmr(400mhz,cdcl3)δ9.56(d,j=3.1hz,1h),6.14(dd,j=10.8,7.2hz,2h),5.65(s,1h),2.59
–
2.44(m,1h),2.44
–
2.29(m,3h),2.25(ddd,j=13.1,5.3,1.9hz,1h),2.01
–
1.70(m,7h),1.70
–
1.31(m,6h),1.31
–
1.20(m,4h),1.12(d,j=6.9hz,3h),0.78(s,3h).
[0193]
13
c nmr(101mhz,cdcl3)δ204.51(s),199.36(s),163.08(s),140.68(s),126.90(s),123.70(s),50.79(s),49.34(s),48.93(s),43.21(s),39.72(s),38.58(s),38.32(s),37.10(s),35.51(s),33.88(s),26.66(s),25.27(s),22.13(s),20.47(s),13.28(s),11.00(s).
[0194]
质谱:c21h28o2,313.0。
[0195]
以下实施例提供地屈孕酮的合成路线中部分环节的其他合成方法。实施例7-9的路线如下:
[0196][0197]
实施例7
[0198][0199]
在500ml的三口烧瓶里50g(0.10mol)化合物b和200ml dmf,溶清,加50g(0.51mol)醋酸钾,100℃反应2小时,tlc检测原料反应完全,缓慢倒入1l水中,析出固体,搅拌1小时,过滤,烘干得黄色固体35g,摩尔收率约92%。
[0200]
经检测:1h nmr(400mhz,cdcl3)δ5.64(dd,j=5.3,2.5hz,1h),5.52
–
5.39(m,1h),4.06(dd,j=10.8,3.4hz,2h),3.78(dd,j=10.7,7.4hz,1h),2.59
–
2.39(m,2h),2.25(dt,j=15.7,2.6hz,2h),2.03(s,3h),1.94(dd,j=8.6,3.8hz,2h),1.75
–
1.59(m,7h),1.42(ddd,j=29.8,15.4,6.9hz,6h),0.98(d,j=6.6hz,3h),0.71(s,3h),0.61(s,3h).
[0201]
实施例8
[0202][0203]
在1l的三口烧瓶里加78g(0.21mol)化合物b2,水3.78g(0.21mol),碳酸氢钠35g(0.41mol)和500ml二氯甲烷,5℃~10℃加戴斯马丁试剂105g(0.28mol),保温搅拌半小时,tlc显示原料反应生成中间态异构体,降温-20℃冷冻1~2小时,过滤,滤饼用冷二氯甲烷适量淋洗至滤饼无产物,有机相依次用亚硫酸钠溶液和碳酸氢钠溶液,食盐水洗涤。
[0204]
有机相加100ml三乙胺,常温搅拌1-2小时,tlc显示中间态异构体转化为化合物c1,有机相依次用食盐水,1m稀盐酸,食盐水洗涤,有机相40℃浓缩,正庚烷置换至小体积,降温-20℃冷冻1-2小时,过滤,烘干得黄色固体51g,摩尔收率约65%。
[0205]
经检测:1h nmr(400mhz,cdcl3)δ5.79(s,1h),5.25
–
5.16(m,1h),4.05(dd,j=10.7,3.4hz,1h),3.77(dd,j=10.7,7.3hz,1h),3.62
–
3.54(m,1h),3.15
–
2.86(m,2h),2.44
–
2.36(m,1h),2.25
–
2.17(m,1h),2.03(s,3h),1.87
–
1.67(m,7h),1.54
–
1.49(m,2h),1.38(d,j=11.2hz,2h),1.26
–
1.23(m,2h),1.03(s,3h),0.98(d,j=6.6hz,3h),0.65(s,3h).
[0206]
实施例9:
[0207][0208]
在1l的三口烧瓶里加840ml的无水乙醇,低温通入干燥的氯化氢气体,制备得无水乙醇/氯化氢溶液(水分要小于0.2%,含量约35%);在2l的三口烧瓶里,加54g(0.146mol)化合物c1,0.54g tbhq和700ml二氯甲烷,溶清,氮气保护,0℃~10℃下,滴加840ml自制的无水乙醇/氯化氢溶液,控温反应1小时左右,tlc检测原料剩余小于3%,加纯净水淬灭反应,分液,有机相用碳酸氢钠溶液洗至ph=7-8,有机相50℃以下浓缩,乙醇置换,保留约500ml乙醇,边搅拌边梯度降温,黄色固体析出,降温-20℃冷冻1-2小时,过滤,粗品再用正庚烷打浆,冷析过滤,烘干得米白色固体36g,摩尔收率约66.7%。
[0209]
经检测:1h nmr(400mhz,cdcl3)δ6.15(dt,j=19.5,7.4hz,2h),5.64(s,1h),4.07(dd,j=10.7,3.5hz,1h),3.76(dd,j=10.7,7.3hz,1h),2.59
–
2.44(m,1h),2.40(ddd,j=11.5,7.4,4.9hz,2h),2.24(ddd,j=13.1,5.2,1.9hz,1h),2.03(s,3h),1.95
–
1.67(m,7h),1.67
–
1.52(m,2h),1.43
–
1.31(m,2h),1.29
–
1.13(m,5h),1.00(d,j=6.6hz,3h),0.75(s,3h)。
[0210]
13
c nmr(101mhz,cdcl3)δ199.40(s),171.22(s),163.31(s),141.05(s),126.73(s),123.59(s),69.30(s),52.66(s),49.32(s),42.80(s),39.68(s),38.73(s),38.45(s),37.13(s),35.61(d,j=13.6hz),33.90(s),27.21(s),25.01(s),22.14(s),20.91(s),
20.53(s),16.99(s),10.66(s)。
[0211]
质谱:c
24h34
o3,371.0。
[0212]
实施例10-13的路线如下:
[0213][0214]
实施例10:
[0215][0216]
在500ml的三口烧瓶里50g(0.10mol)化合物b和200ml dmf,溶清,加17.4g(0.21mol)溴化锂,100℃反应2小时,tlc检测原料反应完全,缓慢倒入1l水中,析出固体,搅拌1小时,过滤,烘干得黄色固体37.7g,摩尔收率约93%。
[0217]
实施例11:
[0218][0219]
在1l的三口烧瓶里加82g(0.21mol)化合物b3,3.78g水(0.21mol),碳酸氢钠35g(0.41mol)和500ml二氯甲烷,5℃~10℃加戴斯马丁试剂105g(0.28mol),保温搅拌半小时,tlc显示原料反应生成中间态异构体,降温-20℃冷冻1-2小时,过滤,滤饼用冷二氯甲烷适量淋洗至滤饼无产物,有机相依次用亚硫酸钠溶液和碳酸氢钠溶液,食盐水洗涤。
[0220]
有机相加100ml三乙胺,常温搅拌1-2小时,tlc显示中间态异构体转化为化合物bbr,有机相依次用食盐水,1m稀盐酸,食盐水洗涤,有机相40℃浓缩,正庚烷置换至小体积,降温-20℃冷冻1-2小时,过滤,烘干得黄色固体55.8g,摩尔收率约68%。
[0221]
实施例12:
[0222][0223]
在1l的三口烧瓶里加840ml的无水乙醇,低温通入干燥的氯化氢气体,制备得无水乙醇/氯化氢溶液(水分要小于0.2%,含量约35%);在2l的三口烧瓶里,加57g(0.146mol)化合物c2,0.57g tbhq和700ml二氯甲烷,溶清,氮气保护,0℃~10℃下,滴加840ml自制的无水乙醇/氯化氢溶液,控温反应1小时左右,tlc检测原料剩余小于3%,加纯净水淬灭反应,分液,有机相用碳酸氢钠溶液洗至ph=7-8,有机相50℃以下浓缩,乙醇置换,保留约500ml乙醇,边搅拌边梯℃降温,黄色固体析出,降温-20℃冷冻1-2小时,过滤,粗品再用正庚烷打浆,冷析过滤,烘干得固体38g,摩尔收率约66.7%。
[0224]
经检测:1h nmr(400mhz,cdcl3)δ6.30
–
6.04(m,2h),5.66(s,1h),3.50(dd,j=9.8,2.5hz,1h),3.36(dd,j=9.8,5.7hz,1h),2.64
–
2.48(m,1h),2.47
–
2.34(m,2h),2.32
–
2.21(m,1h),1.95
–
1.55(m,9h),1.47
–
1.17(m,7h),1.09(d,j=6.5hz,3h),0.77(s,3h).
[0225]
实施例13:
[0226][0227]
在250ml的三口烧瓶里加90ml甲醇和40ml二氯甲烷以及30g(76.6mmol)化合物d2,0℃~10℃下滴加61ml氢氧化钠水溶液(10%,153.2mmol),控温反应1小时左右,tlc检测原料反应完,加10ml醋酸淬灭反应,有机相50℃以下浓缩除去二氯甲烷,甲醇水出料,冷析过滤,烘干得固体22.7g,摩尔收率约90%。
[0228]
经检测:1h nmr(400mhz,cdcl3)δ6.17(dt,j=23.1,7.5hz,2h),5.65(s,1h),3.64(dd,j=10.5,3.2hz,1h),3.39(dd,j=10.5,6.6hz,1h),2.69
–
2.46(m,1h),2.46
–
2.32(m,2h),2.25(ddd,j=13.1,5.2,1.9hz,1h),1.82(dddd,j=16.7,14.9,13.7,9.7hz,6h),1.69
–
1.50(m,4h),1.38(ddd,j=17.9,12.4,4.6hz,2h),1.28
–
1.15(m,5h),1.05(d,j=6.6hz,3h),0.76(s,3h)。
[0229]
13
c nmr(101mhz,cdcl3)δ199.62(s),163.54(s),141.31(s),126.72(s),123.56(s),67.78(s),52.27(s),49.36(s),42.72(s),39.73(s),38.93
–
38.34(m),37.17(s),35.55(s),33.93(s),27.32(s),25.05(s),22.15(s),20.57(s),16.65(s),10.70(s)。
[0230]
质谱:c22h32o2,329.0。
[0231]
实施例14:
[0232][0233]
在500ml的三口烧瓶里加200ml二氯甲烷50g(0.10mol)化合物b和13.6g(0.20mol)咪唑,-5℃~5℃下滴加16.3g三甲基氯硅烷(0.15mol),控温反应1小时左右,tlc检测原料反应完,加10ml水淬灭反应,有机相依次用水,稀盐酸水溶液,碳酸氢钠溶液洗涤,有机相50℃以下浓缩除去二氯甲烷,乙醇置换出料,冷析过滤,烘干得固体51.7g,摩尔收率约90%。
[0234]
b4采用类似实施例7的方法酯化后,脱保护得到b2。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1