眼肿瘤融合基因的检测模型及构建方法和检测方法与流程
1.本发明涉及体外诊断检测技术领域,具体涉及一种眼肿瘤融合基因的检测模型及构建方法和检测方法。
背景技术:
2.融合基因是指两个基因全部或一部分相互融合为一个新基因的过程,是染色体异位、中间缺失或染色体倒置所致,通常具有致瘤性,在各种不同的肿瘤中普遍存在。融合基因是肿瘤的普遍特征,并可作为肿瘤分子的诊断和靶标。融合基因引发癌症的机制主要有两种:一部分发生在肿瘤驱动基因上的基因融合使这些基因拥有强的启动子或增强子,或激活转录因子,使其异常表达;另一部分融合基因会产生有生物学作用(如激酶活性等)的嵌合体蛋白,使生物体紊乱。可用药的激酶融合在检测的单个癌症类型中检测到的频率为1%-9%,包括alk、ros1、ret、ntrks和fgfr等。目前针对融合基因的肿瘤靶向药物已有多种获得fda/cfda批准,如针对alk、ros1基因融合的克唑替尼,以及针对ntrks基因融合的larotrectinib等。美国国家综合癌症网络(nccn)指出,含有融合基因的患者可接受相应的靶向药物治疗,推荐的检测手段有高通量测序(ngs)、实时荧光定量pcr(rt-pcr)、荧光原位杂交(fish),以及免疫组化(ihc)等。
3.由于fish和ihc等方法具有通量低、检测周期长、费用昂贵等缺点,而rt-pcr只能用来检测已知融合变异,ngs技术被越来越多用在肿瘤基因融合的检测上。基于此,开发一种更为简便、成本低和样本投入少的肿瘤融合基因检测方法。
技术实现要素:
4.针对上述现有技术的不足,本发明所要解决的问题是:如何提供一种眼肿瘤融合基因的检测模型及构建方法和检测方法,以解决现有技术中对肿瘤融合基因检测操作复杂、成本高和样本投入大的问题。
5.为了解决上述问题,本发明采用了如下的技术方案:
6.一种构建基于转录组rna-seq的眼肿瘤融合基因检测模型的方法,包括:
7.(1)基于融合基因设计探针;
8.(2)从离体组织样本或血液中提取总rna,并进行总rna浓度和质量控制;
9.(3)将提取的所述总rna用磁珠富集真核生物mrna或用试剂盒去除原核生物rrna,加入fragmentation buffer将mrna打断成短片段,以mrna为模板,用六碱基随机引物合成第一条cdna链,然后加入缓冲液、dntps、rnase h和dna polymerase i合成第二条cdna链,在经过qiaquick pcr试剂盒纯化并加eb缓冲液洗脱之后,进行末端修复、加polya并连接测序接头,然后用琼脂糖凝胶电泳进行片段选择,经pcr扩增后,进行文库质检,获得dna文库;
10.(4)将所述dna文库用进行靶向捕获,得到靶向捕获文库并进行质检,得测序文库,将所述测序文库用illumina hiseq进行测序;
11.(5)将illumina hiseq测序得到原始图像数据文件经碱基识别分析转化为原始测
序序列,将所述原始测序序列去除接头和低质量碱基;将处理后的原始测序序列进行分析,并与所述原始测序序列相应的参考基因组信息进行对比。
12.进一步,所述步骤(2)总rna浓度和质量控制包括总rna浓度≥20ng/ul、总rna质量2.1≥od
260/280
≥1.9。
13.进一步,所述步骤(4)中测序文库浓度≥5ng/ul。
14.进一步,所述步骤(1)中融合基因包括abcb9、abl1、ackr3、acsl3、actb、adgrg7、aff3、ahrr、akap12、akap9、aldh2、alk、aspscr1、atf1、atic、bag4、baiap2l1、bcor、bcr、braf、brd3、brd4、c11orf95、camta1、cant1、cars、casp7、catsperz、ccar2、ccdc6、ccnb1ip1、ccnb3、ccnd1、ccnd3、cdh11、cdkn1a、cdx1、chchd7、chn1、cic、ciita、cltc、cnbp、col12a1、col1a1、col1a2、col4a5、col6a3、cox6c、creb1、creb3l1、creb3l2、crtc1、crtc3、csf1、csf1r、ctnnb1、cxorf67、dctn1、ddit3、ddx5、dleu2、dux4、ebf1、egfr、eif3e、eif4a2、eml4、elk4、ep300、epc1、epcam、erbb2、erbb4、erc1、erg、erlin2、esr1、esrra、etv1、etv4、etv5、etv6、ewsr1、ezr、fev、fgf8、fgfr1、fgfr2、fgfr3、fhit、fip1l1、fli1、fn1、foxo1、foxo4、fus、gli1、golga5、gopc、gpc3、has2、herpud1、hey1、hjurp、hmga1、hmga2、hmgn2p46、hnrnpa2b1、hook3、hpr、irf2bp2、jazf1、kdr、kiaa1549、kif5b、kit、klf17、klk2、klk4、klkp1、kras、ktn1、lgr5、lhfpl6、lifr、lmo1、lpp、lrig3、lrp1、maml2、mbtd1、meaf6、met、cd74、mipol1、mn1、mrtfb、msh2、mutyh、myb、myc、nab2、ncoa1、ncoa2、ncoa4、ndrg1、nfatc1、nfatc2、nfib、nono、notch1、notch2、nr4a3、nrg1、ntrk1、ntrk2、ntrk3、nup107、nutm1、nutm2a、nutm2b、nutm2e、oga、omd、pafah1b2、patz1、pax3、pax7、pax8、pbx1、pbx3、pcm1、pcsk7、pdgfb、pdgfra、pdgfrb、phf1、pik3ca、plag1、plpp3、pou5f1、pparg、ppfibp1、prcc、prkar1a、pspc1、ptgfrn、ptprk、rad51b、raf1、ranbp2、relch、ret、rgs17、rmi2、ros1、rps10、rspo2、rspo3、runx1、runx1t1、sdc4、sdha、sdhb、sdhd、sec16a、sec22b、sec31a、sept14、sfpq、slc34a2、slc45a3、slc49a4、smarca5、smarcb1、smarce1、sox9、sp3、srgap3、ss18、ss18l1、ss18l2、ssx1、ssx2、ssx4、stat6、suz12、tacc1、tacc3、taf15、tbl1xr1、tcea1、tcf12、tcf7l2、tec、tenm4、tert、tet1、tfe3、tfeb、tfg、tgfbr3、thrap3、tmprss2、tp53、tp53bp1、tpm3、tpm4、tpr、trim24、trim27、trim33、usp6、vti1a、wasf2、wdcp、wif1、wt1、wwtr1、yap1、ywhae、yy1、zc3h7b、zdhhc7、znf331、znf444。
15.进一步,所述步骤(1)设计探针包括:基于所述融合基因,去掉重复区域的序列,从第一个碱基开始截取78bp的序列做探针,再一次往后移动n个碱基,截取78bp的序列做探针,直到最后一个78bp。
16.进一步,所述步骤(5)将处理后的原始测序序列进行分析,并与所述原始测序序列相应的参考基因组信息进行对比包括:分析能定位到基因组上的序列的数量和在参考序列上有多个比对位置的序列的数量。
17.进一步,所述步骤(5)将处理后的原始测序序列进行分析,并与所述原始测序序列相应的参考基因组信息进行对比包括:分析基因的readcount值和基因的fpkm值。
18.进一步,所述步骤(5)将处理后的原始测序序列进行分析,并与所述原始测序序列相应的参考基因组信息进行对比包括:分析junctionreads和spanningfrags的数目。
19.一种由所述构建基于转录组rna-seq的眼肿瘤融合基因检测模型的方法所构建的检测模型。
20.一种基于转录组rna-seq的眼肿瘤融合基因的非疾病诊断的检测方法,利用所述检测模型,将处理后的原始测序序列进行分析,并与所述原始测序序列相应的参考基因组信息进行对比,得出眼肿瘤融合基因的分析结果。
21.本发明的有益效果在于:本发明提供的眼肿瘤融合基因的检测模型及构建方法和检测方法,解决现有技术中对眼肿瘤融合基因检测操作复杂、成本高和样本投入大的问题,并采用临床样本对其进行定位定量的表达验证,寻找其与临床相关性的证据,评价临床价值,探索与早期发现、分类、评价预后相关的眼肿瘤标志物,以及选择更加有效、准确的眼肿瘤治疗靶位奠定研究基础。
附图说明
22.为了使本发明的目的、技术方案和优点更加清楚,下面将结合附图对本发明作进一步的详细描述,其中:
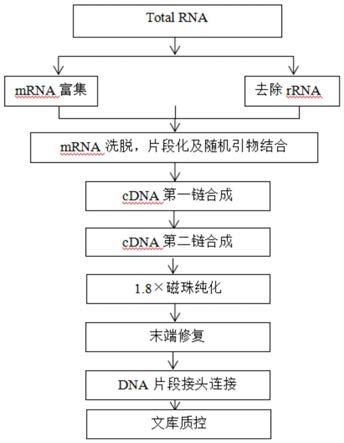
23.图1为本发明方法流程示意图。
具体实施方式
24.下面结合具体实施例对本发明作进一步的详细说明。
25.需要说明的是,这些实施例仅用于说明本发明,而不是对本发明的限制,在本发明的构思前提下本方法的简单改进,都属于本发明要求保护的范围。
26.1、检测融合基因列表
27.[0028][0029]
2、探针设计
[0030]
基于上表中的基因序列,去掉重复区域的序列,重复序列采用repeatmask软件分析得到。从第一个碱基开始截取78bp的序列做探针,再一次往后移动n个碱基,截取78bp的序列做探针,直到最后一个78bp。每个区域根据外显子的gc含量不同,n会有变化,gc含量太高或太低、n越小,探针设计越密集以达到捕获的均一性提高。
[0031]
3、提取样本总rna
[0032]
福尔马林固定和石蜡包埋组织(ffpe):按照mirneasy ffpe kit(qiagen,217504)试剂盒说明书提取总rna。
[0033]
新鲜组织:新鲜组织按照rnapure tissue&cell kit(康为,cw0584s)试剂盒说明书提取总rna.
[0034]
新鲜血液:新鲜外周血样本采用paxgene blood rna tube(bd,762165)采集,按照paxgene blood rna kit(qiagen,762174)试剂盒提取总rna。
[0035]
4、文库构建
[0036]
提取样品总rna后,用带有oligo(dt)的磁珠富集mrna或用rrna探针去除rrna。加入fragmentation buffer将mrna打断成短片段,以mrna为模板,用六碱基随机引物(random hexamers)合成第一条cdna链,然后加入缓冲液、dntps、rnase h和dna polymerase i合成第二条cdna链,在经过qiaquick pcr试剂盒纯化并加eb缓冲液洗脱之后,做末端修复、加polya并连接测序接头,然后用琼脂糖凝胶电泳进行片段大小选择,经pcr扩增后,进行文库质检,浓度≥15ng/ul即为构建成功。流程见附图1。
[0037]
5、文库靶向富集
[0038]
将构建好完成的dna文库用探针进行靶向捕获,捕获参照201811600116.3,得到靶向捕获文库并进行质检,一般认为浓度≥5ng/ul即为构建成功,建好的测序文库使用nextseq500、x ten、novaseq等二代测序平台进行高通量测序,得到测序原始数据。
[0039]
6、生信分析流程
[0040]
获得原始测序序列(sequenced reads)后,并且其相应的基因组参考序列
(reference genome)可以获得的情况下,可以用有参考基因组信息分析流程对数据进行详细的分析:
[0041]
(1)原始序列数据
[0042]
高通量测序(illumina hiseq)测序得到的原始图像数据文件经碱基识别(base calling)分析转化为原始测序序列(sequenced reads),称为raw data或raw reads,结果以fastq(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。fastq格式文件中每个read由四行描述,如下:
[0043]
@st-e00126:486:h555hccxy:7:1101:12672:1221 1:n:0:ngatcgca
[0044]
ntccgcctggaggtcaccgacggccccccggccacccccgcctactgggacggcgagaag
[0045]
+
[0046]
#aaafjjfjjaajajjjfffjffjjjjjjf7a777fjfjjjjf7j7jjfajjjjjfjfff《a
[0047]
其中第一行以“@”开头,随后为illumina测序标识符(sequence identifiers)和描述文字(选择性部分);第二行是碱基序列;第三行以“+”开头,随后为illumina测序标识符(选择性部分);第四行是对应序列的测序质量(cock et al.)
[0048]
(2)测序数据过滤
[0049]
测序所得到的原始测序序列raw reads,里面含有带接头的、低质量的reads,为了保证信息分析质量,必须对raw reads过滤,得到clean reads,后续分析都基于clean reads。样品测序产出数据质量评估情况详见下表1:
[0050]
表1序列质量统计
[0051][0052]
(3)参考序列比对分析
[0053]
在参考基因组选择合适并且相关实验不存在污染的情况下,实验所产生的测序的定位的百分比正常情况下会高于70%(total mapped reads or fragments),其中具有多个定位的测序序列(multiple mapped reads or fragments)占总体的百分比通常不会超过10%。结果见表2:
[0054]
表2样本测序序列与参考基因组的序列比对结果统计
[0055] 2019-rnaseq-001total reads33861316total mapped30892913(91.23%)multiple mapped1424878(4.21%)
[0056]
total reads:序列经过测序数据过滤后的数量统计(clean data)。
[0057]
total mapped:能定位到基因组上的序列的数量的统计;在不存在污染并且参考基因组选择合适的情况下,这部分数据的百分比大于70%。
[0058]
multiple mapped:在参考序列上有多个比对位置的序列的数量统计;这部分数据的百分比一般小于10%。
[0059]
(4)融合基因分析
[0060]
融合基因列表如下所示。其中junctionreads和spanningfrags是检测融合基因的主要指标;在双端测序中,可以把read1和read2看成是一个fragment片段(250bp-300bp)的两端,若read1或read2跨越融合事件的断点,这样的read称为junctionread。read1和read2中间的gap区域横跨融合事件的断点,这样的fragment片段称为spanningfrag,具体分析结构见表3
[0061]
表3融合基因分析结果
[0062][0063]
fusionname:融合事件名称,
[0064]
cosmic:该融合事件是否在cosmic数据库中,如果为yes则可信度很高,
[0065]
junctionreadcount:支持融合事件发生的junction reads数目,
[0066]
spanningfragcount:支持融合事件发生的spanning fragments数目,
[0067]
leftgene:融合事件的上游基因,包括基因id和基因名称,
[0068]
rightgene:融合事件的下游基因。
[0069]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管通过参照本发明的优选实施例已经对本发明进行了描述,但本领域的普通技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离所附权利要求书所限定的本发明的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1