用于前列腺癌的分子分类器的制作方法
用于前列腺癌的分子分类器
1.相关申请
2.本技术要求于2020年6月18日提交的美国临时申请no.63/040,692的优先权,其内容通过引用整体并入。
技术领域
3.本发明涉及分子分类器(molecular classifier),并且更具体地涉及用于前列腺癌的分类器。
背景技术:
4.尽管前列腺癌(cap)是癌症死亡的主要原因,但大多数活检确定病例都是足够惰性的进而安全监测而无需明确治疗[1,2]。侵袭性前列腺癌最有力的生物标志物已格利森分级(gleason grade),这是通过对手术取出的前列腺进行全面病理检查确定的。低格利森级癌症,定义为格利森级3+3=6或who级组(grade group,gg)1[3],表现出可以忽略不计的转移或死亡风险[4,5]。更高级的癌症(who gg2至gg5)需要明确的治疗。与分级方案优先考虑核形态和有丝分裂计数的大多数癌症类型不同,前列腺癌gg只关注腺体结构。良性前列腺和由gg1前列腺癌细胞形成的腺体二者的特征均在于围绕单个腔的单层腔上皮细胞。所有癌细胞都占据相似的环境,在顶端面直接接触腔,在它们的底部有基质,在其余四个侧面有其他癌细胞。这种布置提供了从周围血管中获取氧和营养物的类似途径。相比之下,更高级的癌症(gg2至gg5)形成具有多个腔的融合腺样结构,或者根本不产生腔,这反映了关于细胞间相互作用、分化和代谢方面更强的可塑性。在这些不同布置中生长的能力对应于在前列腺外作为转移性沉积物(metastatic deposit)生长的能力。因此,癌症代谢、上皮可塑性和上皮-基质相互作用是前列腺癌进展的关键主题[6-9]。与gg相关的腺体结构的分子基础为开发用于侵袭性前列腺癌的诊断生物标志物提供了方向。
[0005]
在美国、加拿大和欧洲,主动监测(active surveillance,as)代表了gg1癌症的标准护理[10-13]。患者接受前列腺特异性抗原(prostate-specific antigen,psa)水平和一系列核心活检的监测,并且可能会接受影像作为辅助[10]。虽然基于前列腺切除术的gg是高度信息性的,但目前的方法不能基于针活检准确分离gg1和gg2,呈现为主要困境。由于核心活检的采样误差和观察者间变异性,活检分级在36%至67%的病例中不能准确地反映手术gg[14-17]。这些不准确的后果是男性被置于错误的风险类别中。符合as条件的那些人可能会接受积极的手术干预(根治性前列腺切除术)并遭受过度的发病率,因为与他们患有侵袭性高级癌症的真实风险相关的不确定性。相反,其他人未能及时接受他们需要的治疗以防止无法治愈的转移性疾病的传播。
[0006]
活检中gg的不准确报告激发了分子方法来改善基于cap核心活检采样的风险分层[18]。然而,用于活检gg的现有分子分类器无法准确区分gg1和gg2[19,20]。
[0007]
发明概述
[0008]
在一方面中,提供了在患有前列腺癌的对象中预测疾病进展风险的方法,所述方
法包括:a)提供含有来自肿瘤细胞的rna和dna物质的样品;b)确定或测量以下的值:基本上所有的表6中针对pronto-e列出的353种患者特征,包含mrna和拷贝数畸变(cna)特征,以及一些或所有的表6中所示的参考或对照特征;c)将所述患者特征与参考或对照特征进行比较;以及d)使用将所述患者特征值作为输入的分类器来计算预测评分,所述分类器先前已针对来自早期前列腺癌患者群体的样品进行了训练。
[0009]
在一方面中,提供了在患有前列腺癌的对象中预测疾病进展风险的方法,所述方法包括:a)提供含有来自肿瘤细胞的rna和dna物质的样品;b)确定或测量基本上所有的表6中针对pronto-m列出的94种患者特征,包含mrna、cna、甲基化和临床特征,以及一些或所有的表6中所示的参考或对照特征;c)将所述患者特征与参考或对照特征进行比较;以及d)使用将所述患者特征值作为输入的分类器来计算预测评分,所述分类器先前已针对来自早期前列腺癌患者群体的样品进行了训练。
[0010]
在一方面中,提供了在患有前列腺癌的患者中预测疾病进展风险的计算机实施方法,所述方法包括:a)在至少一个处理器接收反映以下的数据:基本上所有的对应于关于前列腺癌肿瘤的pronto-e或pronto-m分类器的权利要求1或7中限定的患者特征,以及一些或所有的表6中所示的参考或对照特征;b)在至少一个处理器基于患者特征构建患者概况(patient profile);c)在所述至少一个处理器将所述患者概况与参考或对照进行比较;d)在所述至少一个处理器使用将所述患者概况作为输入的分类器来计算预测评分,所述分类器先前已针对来自早期前列腺癌患者群体的样品进行了训练。
[0011]
在一方面中,提供了计算机程序产品,其用于与具有处理器和连接至所述处理器的存储器的通用计算机结合使用,所述计算机程序产品包含其上编码有计算机机制的计算机可读存储介质,其中所述计算机程序机制可以被加载到所述计算机的所述存储器中并使所述计算机执行权利要求13至15中任一项所述的方法。
[0012]
在一方面中,提供了计算机可读介质,其上存储有用于存储根据权利要求16所述的计算机程序产品的数据结构。
[0013]
在一方面中,提供了用于在患有前列腺癌的患者中预测疾病进展风险的装置,所述装置包含:至少一个处理器;和与所述至少一个处理器通信的电子存储器,所述电子存储器存储处理器可执行代码,所述处理器可执行代码在所述至少一个处理器处被执行时使所述至少一个处理器:a)接收反映以下的数据:基本上所有的对应于关于前列腺癌肿瘤的pronto-e或pronto-m分类器的权利要求1或7中限定的患者特征,以及一些或所有的表6中所示的参考或对照特征;b)将所述患者特征与所述参考或对照特征进行比较;以及c)在所述至少一个处理器使用将所述患者概况作为输入的分类器来计算预测评分,所述分类器先前已针对来自早期前列腺癌患者群体的样品进行了训练。
[0014]
附图简述
[0015]
本发明的优选实施方案的这些和其他特征将在其中参考附图的以下详细描述中变得更加明显,其中:
[0016]
图1.方法概述。
[0017]
(a)将病例分为训练组群和验证组群。从每个切除的肿瘤(即每个病例)中提取高级样品和低级样品二者。(b)对与gg相关的431个基因/基因座进行谱分析。(c)使用机器学习流水线(machine learning pipeline)来开发gg分类器。第一,选择了一种或更多种数据
类型。第二,将相关数据划分以进行五倍交叉验证。第三(任选地),丢弃与gg没有显著单变量关联的特征。第四,在选择机器学习算法之后,在四个分区上训练分类器,并在第5个分区上进行测试。
[0018]
图2.来自重复交叉验证的前25个分类器的性能。
[0019]
每列代表一个分类器。顶部图表示分类器使用的数据集、用于训练它的机器学习算法、样品加权(即包络)方案和使用的训练样品类型(见方法)。在auc图中,每个框总结了来自交叉验证1000次重复的平均auc。在gg1和gg≥2图中,每个框分别总结了正确分类的gg1和gg≥2病例的平均分数。平均统计数据根据x
平均值
=(x
低
+x
高
)/2计算,其中x
低
和x
高
分别是仅从低级或高级样品计算的统计数据。分类器按auc降低排序。缩写:auc-曲线下面积(area under the curve);bcr-生物化学复发(biochemical recurrence);capra-前列腺癌风险评估(cancer of the prostaste risk assessment);cn_mlpa-拷贝数,mlpa平台;cn_ns-拷贝数,nanostring平台;gg-分级组(grade group);msp-甲基化特异性pcr(methylation-specific pcr)。
[0020]
图3.多模态分类器pronto-e和pronto-m的性能。
[0021]
(a至c)多模态分类器,即使用不同类型数据的分类器,在交叉验证中优于单模态分类器。每个分类器的tp率(a)、fp率(b)和auc(c)从重复1000次的交叉验证计算得出(框总结了重复)。在每次重复中,每个统计数据使用来自每个病例的仅高级或低级样品来计算。高级和低级统计数据的平均值显示在“平均值”部分。给定分类器使用的输入数据类型在(c)中的图例中指示;capra仅使用临床数据。根据交叉验证,多模态分类器是性能最好的分类器。(d)多模态分类器的验证性能。对于验证组群中的每个病例,随机选择一个样品,并使用代表性样品计算统计数据。该过程重复1000次,并且每个点表示重复的中位数(即基于采样的auc),并且上下误差条分别表示第一和第三四分位数。(a至c)cna是指来自mlpa的cna数据,因为pronto-e和pronto-m仅使用来自mlpa的cna数据。(e)来自同一验证病例的低级和高级样品的预测类别之间的一致性。(f)在具有一致性的那些病例中,预测正确的百分比。(e至f)gg1病例与gg≥2患者分开。用于计算每个百分比的验证病例总数显示在条形上方。注意,pronto-e和pronto-m的数字不同,因为分类器对每个样品具有不同的数据要求。
[0022]
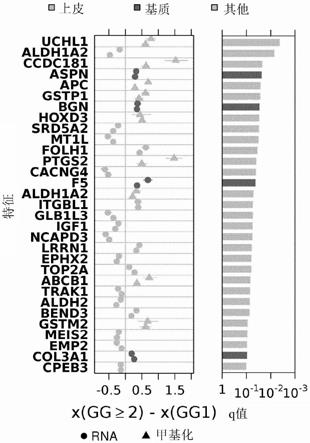
图4.与gg具有显著单变量关联的分子特征(q值<0.1)。
[0023]
对于每个显著的分子特征,左图示出了gg≥2和gg1病例的特征值的中位数差异。示出了每个组群的差异,其中点表示中位数,并且相贯线(intersecting line)的末端表示第一和第三四分位数,每个病例的一个代表性样品的1000个随机选择。右图表示由训练和验证组群q值的组合产生的q值(即经调整的p),表示特征与gg之间单变量关联的显著性(见方法)。mrna特征分析使用了332个训练病例和200个验证病例,并且甲基化特征分析使用了318个训练病例和202个验证病例。对于靶向基因,表明在上皮或基质区室中优先表达[54]。
[0024]
图5.用于植入所述方法的计算机装置。
[0025]
适当配置的计算机装置和相关的通信网络、装置、软件和固件,以提供用于实现如本文中所述的一个或更多个实施方案的平台。
[0026]
图6.gg分类器设计概述。
[0027]
gg分类器将患者概况(profile)作为输入,其中概况可能包含不同数据类型的特征(包括临床特征,未示出)。将分类器使用数种可能的机器学习算法之一进行训练(见方
法),以预测患者是否患有病理gg≥2。也就是说,最终的分类器输出将是是或否。
[0028]
图7.不同操作点处的pronto-e和pronto-m。
[0029]
(a)pronto-e和pronto-m分类器针对来自每个病例的仅低级或仅高级样品的验证roc曲线。预测评分是分类器的数值输出,并且操作点为x,评分>=x预测病理gg>=2,而评分<x预测病理gg1。曲线示出了不同操作点的真阳性率和假阳性率。(b)pronto-e和pronto-m分类器的预测评分分布。框表示来自应用于验证组群中所有样品的分类器的评分分布,由其源病例的gg分隔。正如预期的那样,对于两个分类器,来自更高gg病例的样品的评分往往更高。红线表示选择的操作点0.5。
[0030]
图8.来自同一病例的低级和高级样品的分子谱之间的相似性。
[0031]
cna指来自mlpa的cna数据,因为pronto-e和pronto-m仅使用来自mlpa的cna数据。缩写:methyl.-甲基化。
[0032]
图9.pronto-e的潜在临床影响。
[0033]
如果将pronto-e分类器应用于推荐进行主动监测的1000名患者的诊断活检中的假设性能。鉴于1000名主动监测患者和pronto-e的预测性能,该图示出了真阳性和假阳性、真阴性和假阴性的假设数目,以及这些患者子集将如何受到他们的测试结果的影响。阳性测试结果将在诊断之后3或6个月触发早期活检,这可能导致升级和后续治疗。相反,阴性测试结果将导致在诊断之后12个月进行活检。
[0034]
发明详述
[0035]
在以下描述中,阐述了许多具体细节以提供对本发明的透彻理解。然而,应当理解,可以在没有这些具体细节的情况下实施本发明。
[0036]
癌症分级是早期前列腺癌(cap)中疾病进展的最有力预测指标。瘤内异质性和观察者间变异性限制了诊断活检的准确性,并降低了临床效用。使用前列腺切除术的病理检查作为金标准,我们开发并验证了前列腺癌分级的稳健客观生物标志物。
[0037]
从低风险和中风险cap患者中收集根治性前列腺切除术,并将其分配到训练(n=333)或验证(n=202)组群。为了整合瘤内异质性,每个病例分别在两个位置采样。我们谱分析了富集cap代谢、基质信号传导和上皮可塑性的342个mrna,辅以100个拷贝数畸变(copy number aberration,cna)和14个dna高甲基化基因座。使用训练数据产生了病理级别组(1与≥2)的超过41,000个候选分类器,使临床、病理和分子变量接受12种不同的机器学习算法。我们通过对具有更高真阳性(true positive,tp)率和接受者-操作者曲线下面积(auc)的分类器进行优先排序而选择了两个分类器,pronto-e和pronto-m,以进行验证。
[0038]
pronto-e分类器包含353种mrna和cna特征,而pronto-m分类器包含94种mrna、cna、甲基化和临床特征。分类器(pronto-e、pronto-m)独立验证,真阳性率分别为0.802和0.810,假阳性率分别为0.403和0.398,并且auc分别为0.799和0.786。
[0039]
在不同组群中开发和验证了两个多基因分类器,每个分类器都通过整合不同类型的基因组数据实现了优异的性能。分类器的采用可以在不提高患者发病率的情况下改进当前的主动监测方法。
[0040]
在一方面中,提供了在患有前列腺癌的对象中预测疾病进展风险的方法,所述方法包括:a)提供含有来自肿瘤细胞的rna和dna物质的样品;b)确定或测量以下的值:基本上所有的表6中针对pronto-e列出的353种患者特征,包含mrna和拷贝数畸变(cna)特征,以及
一些或所有的表6中所示的参考或对照特征;c)将所述患者特征与参考或对照特征进行比较;以及d)使用将所述患者特征值作为输入的分类器来计算预测评分,所述分类器先前已针对来自早期前列腺癌患者群体的样品进行了训练。
[0041]
在一些实施方案中,基本上所有353种患者特征是所有353种患者特征。
[0042]
本文中使用的术语“对照”是指可用于预测或分类值,例如患者特征包括从与结果类别相关的受试样品获得的mrna、拷贝数畸变(cna)特征或临床特征的特定值或数据集。本领域技术人员将理解受试样品与对照之间的比较将取决于所使用的对照。
[0043]
本文中使用的关于癌症的术语“低风险”或“低可能性”是指与一般或对照群体相比具有统计学上显著性较低的癌症风险。相应地,本文中使用的关于癌症的“高风险”或“高可能性”是指与一般或对照群体相比具有统计学上显著更高的癌症风险。
[0044]
本文中使用的术语“样品”是指来自对象的可测定本文中提及的dna或rna物质的任何流体、细胞或组织样品。
[0045]
在一方面中,提供了在患有前列腺癌的对象中预测疾病进展风险的方法,所述方法包括:a)提供含有来自肿瘤细胞的rna和dna物质的样品;b)确定或测量基本上所有的表6中针对pronto-m所列的94种患者特征,包括mrna、cna、甲基化和临床特征,以及一些或所有的表6中所示的参考或对照特征;c)将所述患者特征与参考或对照特征进行比较;以及d)使用将所述患者特征值作为输入的分类器来计算预测评分,所述分类器先前已针对来自早期前列腺癌患者群体的样品进行了训练。
[0046]
在一些实施方案中,基本上所有94种患者生物标志物是所有94种患者生物标志物。
[0047]
在一些实施方案中,确定预测评分包括将患者肿瘤分类为病理格利森级别组(gg)级别。
[0048]
在一些实施方案中,如果评分≥0.5,则将所述患者肿瘤分类为病理gg≥2级,或者如果评分<0.5,则将所述患者肿瘤分类为病理gg1级。
[0049]
在一些实施方案中,如果将患者分类为病理gg1级,则还包括用主动监测来管理所述患者。在一些实施方案中,如果将患者分类为病理gg≥2级,则还包括用手术、内分泌治疗、化学治疗、放射治疗、激素治疗、基因治疗、热治疗或超声治疗来治疗患者。
[0050]
本发明系统和方法可以以多个实施方案实施。适当配置的计算机装置以及相关的通信网络、装置、软件和固件可以提供用于实现如上所述的一个或更多个实施方案的平台。作为实例,图5示出了通用计算机装置100,其可以包含连接至存储单元104和随机存取存储器106的中央处理单元(“cpu”)102。cpu 102可以处理操作系统101、应用程序103和数据123。可以根据需要,操作系统101、应用程序103和数据123可存储在存储单元104中并加载到存储器106中。计算机装置100还可以包含图形处理单元(gpu)122,其可操作地连接至cpu 102和存储器106以从cpu 102卸载密集的图像处理计算并且与cpu 102并行运行这些计算。操作者107可以使用通过视频接口105连接的视频显示器108、以及通过i/o接口109连接的多个输入/输出装置(例如键盘115、鼠标112和磁盘驱动器或固态驱动器114)与计算机装置100交互。以已知方式,鼠标112可以被配置成控制光标在视频显示器108中的移动,并使用鼠标键来操作出现在视频显示器108中的多种图形用户界面(graphical user interface,gui)控件。磁盘驱动器或固态驱动器114可以被配置成接受计算机可读介质116。计算机装
置100可以通过网络接口111形成网络的一部分,从而允许计算机装置100与其他适当配置的数据处理系统(未示出)通信。一种或更多种不同类型的传感器135可用于接收来自多种来源的输入。
[0051]
本发明系统和方法可以在几乎任何形式的计算机装置上实施,包括台式计算机、膝上型计算机、平板计算机或无线手持式计算机。本发明系统和方法还可以实现为计算机可读/可用介质,所述计算机可读/可用介质包含计算机程序代码以使一个或更多个计算机装置能够实现根据本发明的方法中的多个处理步骤中的每一个。在多个计算机装置执行整个操作的情况下,计算机装置联网以分配多个操作步骤。应当理解,术语计算机可读介质或计算机可用介质包含任何类型的程序代码物理实施方案中的一个或更多个。特别地,计算机可读/可用介质可以包含在一个或更多个便携式存储制品(例如,光盘、磁盘、磁带等)上、在计算机装置中分配的一个或更多个数据存储单元(例如与计算机和/或存储系统缔合的存储器)上体现的程序代码。
[0052]
在一方面中,提供了在患有前列腺癌的患者中预测疾病进展风险的计算机实施方法,所述方法包括:a)在至少一个处理器接收反映以下的数据:基本上所有的对应于关于前列腺癌肿瘤的pronto-e或pronto-m分类器的权利要求1或7中限定的患者特征,以及一些或所有的表6中所示的参考或对照特征;b)在至少一个处理器基于所述患者特征构建患者概况;c)在所述至少一个处理器将所述患者概况与参考或对照进行比较;d)在所述至少一个处理器使用将所述患者概况作为输入的分类器来计算预测评分,所述分类器先前已针对来自早期前列腺癌患者群体的样品进行了训练。
[0053]
在一方面中,提供了计算机程序产品,其用于与具有处理器和连接至所述处理器的存储器的通用计算机结合使用,所述计算机程序产品包含其上编码有计算机机制的计算机可读存储介质,其中所述计算机程序机制可以被加载到所述计算机的所述存储器中并使所述计算机执行如权利要求13至15中任一项所述的方法。
[0054]
在一方面中,提供了计算机可读介质,其上存储有用于存储根据权利要求16所述的计算机程序产品的数据结构。
[0055]
在一方面中,提供了用于在患有前列腺癌的患者中预测疾病进展风险的装置,所述装置包含:至少一个处理器;和与所述至少一个处理器通信的电子存储器,所述电子存储器存储处理器可执行代码,所述处理器可执行代码在所述至少一个处理器处被执行时使所述至少一个处理器:a)接收反映以下的数据:基本上所有的对应于关于前列腺癌肿瘤的pronto-e或pronto-m分类器的权利要求1或7中限定的所述患者特征,以及一些或所有的表6中所示的参考或对照特征;b)将所述患者特征与所述参考或对照特征进行比较;以及c)在所述至少一个处理器,使用将所述患者概况作为输入的分类器来计算预测评分,所述分类器先前已针对来自早期前列腺癌患者群体的样品进行了训练。
[0056]
以下实施例进一步举例说明了本发明的优点。本文中所示的实施例和它们的具体细节仅出于举例说明呈现并且不应被解释为对本发明的权利要求书的限制。
实施例
[0057]
材料和方法
[0058]
患者样品:
[0059]
为了训练和验证分类器,使用金斯敦综合医院(kingston general hospital)(1999年至2012年诊断)、麦吉尔大学健康中心蒙特利尔综合医院(montreal general hospital at mcgill university health centre)(1994至2013年)和伦敦健康科学中心(london health sciences centre,lhsc)(2004至2009)的当地电子病历确定了根治性前列腺切除术样品。最初的纳入标准是(i)在核心活检中审查了gg1或gg2的诊断,(ii)接受了根治性前列腺切除术,以及(iii)手术前未接受过治疗(treatment-)。临床阶段为t3或更高的患者被排除在外。将病例分配到训练群组或验证群组。
[0060]
对于所有病例,诊断核心活检和根治性前列腺切除术二者的中心病理学审查均由专家病理学家(fb、mm、db、tj)进行。在可以的情况下,从从主要肿瘤病灶(dominant tumor focus)的两个区域(图1a)获得的穿刺核心(punch core)中提取dna和rna[21],所述两个区域富含存在的相对高和低gg区域,并使用针对该方法优化的方案[22,23]。进行的所有分析都得到了当地伦理审查小组的批准(表3),其允许豁免知情同意。总体而言,我们从333个病例中收集了633个样品用于训练集,并从202个病例中收集了346个样品用于验证集(见表4中的consort数据)。
[0061]
训练和验证组群的临床病理特征总结在表1中。假设真阳性(tp)率≥0.8并且假阳性(fp)率≤0.55,我们以89%的能力验证了两个分类器(α=0.01)[24]。
[0062]
分类器候选特征的选择:
[0063]
将反映gg生物学的多个功能方面在转录组(mrna丰度)、基因组(dna拷贝数改变(copy number alteration,cna))和表观基因组水平(dna甲基化)的分子特征上进行了探询(图1b)。在详细的文献回顾和研究团队成员领导的数个研究链的输入[25-30]之后,汇总了评估431个基因/基因座(其中一个基因/基因座可由多于一种特征评估)的462种分子特征列表(见方法;表6)。我们还包括了在诊断时评估的四种临床特征,以及将它们整合到前列腺癌风险评估(capra)风险组中的第五种临床特征[31]。我们总共使用了467种特征来描述肿瘤样品(表6)
[0064]
集中的分子谱分析(profiling):
[0065]
我们采用了四个分子诊断平台,其中三个目前在临床上用于癌症的分子诊断。使用nanostring n-counter平台[32]用为本研究开发的特定代码集进行mrna分析。使用专门为此项目开发的基于多重连接依赖性探针扩增(multiplex ligation-dependent probe amplification,mlpa)的测定和定制的nanostring拷贝数代码集[33][34]二者进行cna分析。(ebrahimizadeh et al,提交的手稿)。最后,使用甲基化特异性聚合酶链反应(polymerase chain reaction,msp)[26]进行表观遗传谱分析。鉴于两个组群的rna和dna产量,将两个组群中的所有样品都在尽可能多的平台上进行了分析。
[0066]
预后分类器的开发和验证:
[0067]
如补充方法中所述,对训练和验证数据二者都进行了预处理。我们创建了有监督的机器学习流水线(图1c;补充方法)来开发分类器,所述分类器将患者概况(包含特征值)作为输入,并使用病理前列腺切除术gg作为终点,其中gg1和gg≥2病例分别为阴性和阳性的金标准。使用训练数据,通过在五倍交叉验证中对选定的特征进行12种不同的机器学习算法来评价>41,000个gg分类器。具体来说,计算每个分类器的接受者-操作者曲线下面积(auc)、tp、fp和真阴性(tn)率。这组指标使用来自每个病例的仅低级或高级样品计算,并且
我们还计算了低级和高级统计数据的平均值。我们通过优先排序来自交叉验证中具有更高tp率和auc的分类器来选择两个分类器用于验证。
[0068]
我们通过计算上述统计数据来验证分类器,并且还通过在验证组群中随机选择每位患者的一个样品(高级或低级)来计算性能统计数据来验证分类器,并且重复该过程1000次。这些基于采样的统计数据更好地模拟了临床实践。所有统计学分析均使用r软件框架(v3.4.3)[35]、机器学习包mlr(v2.15.0)[36]和绘图包boutroslab.plotting.general(v5.9.8)[37]进行。
[0069]
伦理审查
[0070]
所有研究均根据三理事会政策声明(tri-council policy statement)(tcps2)进行并在每个参与机构的研究伦理委员会对研究方案进行伦理批准之后进行(表3)。
[0071]
特征选择
[0072]
cna特征:mlpa测定
[0073]
开发了多重连接依赖性探针扩增(mlpa)测定来评估先前与前列腺癌(cap;ebrahimizadeh等人,提交的手稿)中的临床结果相关的拷贝数改变(cna;表6)的14个基因座。测定的基因座包括myc致癌基因s[1至3],pten s[4至7],tp53 s[2,8,9],cdkn1b s[10,11]和rb1 s[12,13]肿瘤抑制子,与转移相关的基因座(例如gabarapl2 s[13,14]和pdpk1 s[15,16]),与维持基因组稳定性相关的基因座(例如rwdd3 s[17至20]、gtf2h2 s[21至24]和wrn s[13,25至27]),以及与cap亚型相关的基因:chd1 s[13,28,29]、map3k7s[13,28,30]、nkx3-1s[13]和pdzd2 s[31,32]。
[0074]
cna特征:cpc-gene nanostring测定
[0075]
使用dna cna测定,加拿大前列腺癌基因组网络(canadian prostate cancer genome network,cpc-gene)确定了低至中风险cap患者中基因组改变百分比与降低的生物化学无复发存活之间的关联,并开发了使用cna特征的分类器来预测患者结果s[33]。nanostring cna测定旨在获得这些特征的值s[34],并且在此我们使用所述测定以包括92种cna特征:85个基因座(包括151个基因)和文献中与cap相关的7个另外的基因(表6)。
[0076]
mrna特征:
[0077]
我们通过组合来自以下研究的基因列表产生了mrna丰度基因组(用于nanostring rna测定):
[0078]
mrna特征:cpc-gene
[0079]
cpc-gene进行了来自中风险患者的样品的rna丰度谱分析s[35],并且对这些数据进行的单变量分析鉴定了与不良预后相关的20个基因。这些基因补充了30个基因,所述30个基因用来自taylor et al s[36]的rna数据的预测模型和类似单变量分析来确定。
[0080]
mrna特征:干细胞特征
[0081]
基因列表来源于将四种雄激素受体(androgen receptor,ar)+cap细胞系(lncap、lapc4、cwr22rvl和vcap)“重编程”为干细胞样表型s[37]。对每个细胞系进行的安捷伦(agilent)基因芯片分析揭示了亲本细胞与重编程细胞之间具有显著丰度变化的转录物。然后将这些转录物在细胞系中进行比较,以得出与重编程相关的132个常见变化基因的排序列表。该特征确定了复发、转移和cap特异性死亡的倾向,如s[37]所述。该列表中的前50个基因包含在rna组中。
[0082]
mrna特征:上皮间质转化(epithelial-to-mesenchymal transition,emt)特征
[0083]
使用geo2r程序和benjamini-hochberg方法进行多次测试校正,比较来自在3维培养物(geo#gse19426)中进行侵袭性生长的pc-3、pc-3m、alva-31、rwpe-2-w99细胞系的基因表达数据s[38]以鉴定在四个细胞系中至少三个中失调的1669个基因。这些基因与sabiosciencesqrt-pcr阵列中的emt相关基因交叉引用。使用stringv 9.1和genemania算法s[39,40],将所得33个重叠基因用作网络构建的种子列表。从所得网络中,37个关键基因(包括连接途径的共同节点)包括在rna组中。
[0084]
mrna特征:对上皮生长和分化的基质影响。
[0085]
筛选出如在胚胎前列腺基质s[41-43]中富集的鉴定的318个基因列表,以富集也在癌症相关成纤维细胞中表达的基因,以及与在四个公开可用的数据集s[36,44-46]中的临床和病理终点(复发、cap死亡和格利森评分)的关联。通过在多个数据集中优先排序与分级组(gg)和/或复发相关的基因,创建了80个基因的列表。
[0086]
mrna特征:肿瘤细胞代谢
[0087]
使用stringv9.1和genemania算法,通过连接了固醇调节元件结合蛋白1(sterol regulatory element binding protein,srebp1)、胰岛素生长因子(insulin growth factor,igf)、ar和细胞因子信号传导抑制子1(socs1)的信号传导途径的计算机内基因网络分析鉴定了与cap代谢相关的86个候选基因s[47]。通过nanostring ncounter测定对发现和验证组群上的这些基因进行了表达分析,每个组群包含来自单独肿瘤的32个格利森模式3和32个格利森模式4病灶。使用曼-惠特尼u检验(mann-whitney u test)(p<0.05)的单变量分析鉴定了25个差异表达的基因。
[0088]
mrna特征:前列腺稳态
[0089]
该研究链利用良性前列腺稳态作为通过类固醇激素的生长和分化以及这些途径在cap中失调的模型。代表这项工作主体的转录物包括fer、ptk2、flt1、lyn、src、jak1、jak3、mark3、stat3、stat5a、edf1、wnt11、itgav、itga2和itgv5。
[0090]
甲基化和mrna特征:cpg岛超甲基化
[0091]
从文献中鉴定出cap中具有cpg岛超甲基化的基因(n=14),并如s[48]所述使用甲基化特异性pcr来测定这些基因的dna甲基化,以得到这些甲基化特征的值(表6)。这些基因(除uchl1外)与7个另外的表观遗传修饰和调节基因一起添加至rna组:dnmt1、ezh2、hdac1、hic1、kcnk2、srp14和tert。
[0092]
总之,整理来自这些链中的每一个的基因产生了新的nanostring mrna组,其包含342个基因(见表6)和另外的持家基因(见补充方法)。我们使用nanostring测定来测量每个基因的mrna丰度,从而得出我们mrna特征的值。
[0093]
临床特征
[0094]
前列腺癌风险评估(capra)评分根据五个临床特征来计算:1)诊断时的年龄,2)诊断时以ng/ml计的psa,3)活检gg(即临床gg),4)临床t阶段和5)与癌症相关的活检核心百分比s[49]。患者的capra评分可以继而用于分配capra风险组(低、中、高),并且我们的候选预后分类器任选地使用该组特征。或者,分类器可以直接使用前四种临床特征。如果诊断时的年龄不可获得,我们使用根治性前列腺切除术时的年龄(如果可获得)。如果诊断时psa不可获得,我们使用术前psa(如果可获得)。对于分类器,活检gg1和gg≥2分别表示为0和1。临床
t阶段被简化为两个可能的值,t1和t2,分别表示为0和1,对于分类器。
[0095]
预处理训练和验证数据
[0096]
mrna丰度数据。
[0097]
为了选择使用的归一化方法,我们测试了nanostringnorm r包(v1.1.22;s[50])支持的96种不同方法,通过尝试不同的参数值组合,即背景={无,平均值,平均值.2sd,最大},代码计数={无,总数,geo.平均值},样品含量={无,持家.总数,持家.geo.平均值,总.总数,前.平均值},othernorm={无,排序.标准}。否则,我们使用轮.值=false、取.对数=true和其余参数的默认值。为了评估每种归一化方法,我们使用所得归一化数据计算了数个指标。这些指标包括:
[0098]
1)如果低丰度持家基因的归一化计数显著低于中等丰度持家基因的归一化计数,并且与高丰度基因相比,中丰度基因的归一化计数类似(单侧studentt检验p<0.05),则通过,否则不合格
[0099]
2)动态范围测量为高丰度持家基因的平均归一化计数相对于低丰度持家基因的平均值的百分比提高
[0100]
3)跨盒复制的对照样品的归一化计数之间的一致性,其中值越大表明批次效应越小
[0101]
4)非正态样品的数目,如果样品跨内源基因的归一化计数分布未通过正态性shapiro-wilk检验(fdr调整的q<0.1),则该样品是非正态的
[0102]
5)显著组群协变量的数目,即其中患者来源(金斯敦综合医院/麦吉尔大学健康中心的蒙特利尔医院)是预测归一化计数的线性模型中的显著协变量的基因,其中gg和生物化学复发状态是其他协变量(fdr调整的p<0.1)
[0103]
6)样品的总归一化计数与其源组织块的年龄之间的相关性
[0104]
7)不合格样品的百分比;如果出现以下情况,则样品可能会不合格:
[0105]
a)任何持家基因的归一化计数=0
[0106]
b)在使用归一化计数计算持家基因的z评分之后,任何|z|>5
[0107]
c)如果进行了codecount归一化,归一化因子<0.3或>3
[0108]
d)样品具有离群值背景水平(|z|>5)
[0109]
e)如果进行了samplecount归一化,rna含量值<1
[0110]
f)如果进行了samplecount归一化,样品具有离群值rna含量值(|z|>5)
[0111]
g)缺失内源基因的比例>0.9,其中如果归一化计数≤0则基因缺失
[0112]
仅考虑通过指标1且盒间一致性>0.9和<10%训练样品不合格的方法,我们通过首先通过指标2至7分别排序,并随后采用decor方法(consrank包v2.0.1;s[51])产生的共识排序对方法进行排序。基于这个排序,我们选择了具有背景=无,codecount=无,samplecontent=持家基因.总数,目标值=5000(基于训练数据粗略估计),并且samplecontent=无的归一化方法。
[0113]
mlpa cna数据
[0114]
一个或两个探针靶向每个基因,并且每个受试样品重复测定。对于每个重复,来自每个受试探针的信号除以十个参考探针中的每一个的信号,得到一组七个比率。对于三个参考样品中的至少两个(新鲜的健康女性基因组、正常ffpe肾组织、正常ffpe乳房淋巴结组
织)(promega),当探针的重复比率的95%置信区间超出探针的95%置信区间时,则该探针被认为对cna呈阳性。如果探针对于其两次重复均呈阳性,则所述探针被认为对受试样品呈阳性。如果重复之间存在差异,则探针被认为对cna呈阴性。如果任一重复没有通过品质控制(ebrahimizadeh,提交的手稿),则没有cna状态分配给给定受试样品中的给定探针。如果一个基因的所有探针都是阳性的,则认为所述基因在受试样品中对于cna呈阳性;如果存在差异,则认为所述基因为阴性;否则,不分配cna状态。对于rwdd3、gtf2h2、chd1、map3k7、nkx3-1、wrn、pten、cdkn1b、rb1、gabarapl2和tp53基因仅考虑缺失,而对于myc、pdpk1和pdzd2基因仅考虑增添。
[0115]
nanostring cna数据
[0116]
如前所述s[34]对数据进行预处理。
[0117]
甲基化数据
[0118]cq
值如前所述s[48]计算。对于给定的受试样品t和靶基因g,我们如下计算甲基化水平:
[0119]mt,g,i,j,k,l
=(c
q p,g,i-c
q p,r,j
)-(c
q t,g,k-c
q t,r,l
)
[0120]
其中:
[0121]
p表示与受试样品在同一板上的阳性对照样品,
[0122]
r表示参考序列(alu),并且
[0123]
i、j、k、l表示重复数。
[0124]
然后将归一化甲基化水平定义为:
[0125]mt、g
=中位数
i、j、k、l
(m
t、g、i、j、k、l
)
[0126]
用于开发预后分类器的机器学习流水线
[0127]
我们建立了流水线来详尽评价用于开发预后分类器的不同方法。具体地,所述流水线使用有监督的机器学习方法来开发将患者概况作为输入来预测良好或不良预后(即分别测试阴性和阳性)的分类器。在我们的应用中,我们将前列腺切除术样本中的gg(即病理gg)二值化以定义患者的真实类别:具有仅gg1作为阴性金标准的患者和具有gg≥2作为阳性金标准的患者(补充图1)。
[0128]
所述流水线包含四个主要阶段:1)数据集,2)分区,3)特征约简(feature reduction)和4)交叉验证(图1c)。
[0129]
第一阶段侧重于准备训练数据集。训练数据集包括:按特征矩阵的患者样品(即每一行代表一个患者概况),以及一组真实的类别值,矩阵中的每个样品都有一个值。所述流水线可以获取不同平台产生的输入数据。在我们的应用中,我们有临床/capra、rna丰度、mlpa/nanostring cna和甲基化数据。对于每个平台,此阶段将数据集约简为没有任何缺失数据的样品。如果期望多个平台,则数据集也被约简为具有来自每个目的平台的数据的样品。最后,从数据集中移除不变特征,即在所有剩余样品中具有相同值的特征。
[0130]
第二阶段侧重于对训练数据集进行分区以用于重复交叉验证。根据所期望的选项,数据集被约简为仅低级样品、仅高级样品或每个患者随机选择的样品。默认情况下,此阶段准备重复1000次的五倍交叉验证,并因此该阶段将数据集的1000个分区创建为五个大小相等的子集。对于每个候选分区,首先将每个样品随机分配给五个子集之一。如果分区对于真实类别、生物化学复发状态(在我们的应用中可以与真实类别相关)和样品来源是平衡
的,因为我们的训练样品获自不同的机构(即金斯敦综合医院,麦吉尔大学健康中心的蒙特利尔医院),则分区被保留。具体来说,对于分区中的每一对子集,使用双侧fisher精确检验来检验与每种特征的关联。如果任何潜在关联显著(p<0.05),则产生另一个候选分区,直到获得平衡的分区。
[0131]
第三阶段侧重于特征约简。对于x倍交叉验证,每个分区都启用x个训练子集。在这个阶段,不变的特征(即在所有样品中具有相同值的特征)从每个训练子集中去除。如果期望的话,然后将测试每个剩余特征与真实类别的单变量关联(例如,使用双侧曼-惠特尼u检验)。具有显著关联(例如p<0.01或0.05)的特征被保留。
[0132]
第四阶段使用mlr包v2.15.0 s[52]使用期望的机器学习算法进行重复的x倍交叉验证(图6)。所述算法的选项(括号中为mlr实现标识符)是:决策树(classif.rpart)、灵活判别分析(classif.earth)、用套索(lasso)或弹性网正则化的glm、交叉验证λ(classif.cvglmnet)、k-最近邻(classif.kknn)、线性判别分析(classif.lda)、逻辑回归(classif.logreg)、朴素贝叶斯(classif.naivebayes)、最近收缩质心(classif.pamr)、二次判别分析(classif.qda)、随机森林(classif.ranger)、正则化判别分析(classif.rda)、支持向量机(classif.svm)。无论选择何种算法,重复的交叉验证都是使用未加权的样品进行的(即默认情况下所有样品的权重相等)。
[0133]
对于支持样品加权的算法,该阶段还交叉验证了阴性/阳性金标准类别的不同权重:30%/70%、40%/60%、50%/50%、60%/40%、70%/30%。具体来说,使用wn%/(100-wn)%加权,每个阴性样品和阳性样品分别被分配wn/pn和(100-wn)/(1-pn)的权重,其中pn是样品在阴性金标准类别的比例;因此,所有阴性样品的总权重占综合总计的wn%,并且所有阳性样品的总权重占综合总计的(100-wn)%。对于所有其他机器学习算法参数,使用默认值。
[0134]
在交叉验证期间,使用给定的机器学习算法、数据集(在早期阶段准备)和样品权重在x倍的(x-1)上训练分类器。如果该训练在3次尝试之后失败,则流水线将跳至使用下一个(x-1)倍数据进行训练。如果成功的话,则从两个角度在剩余的数据倍数上测试所得分类器:i)来自每个病例的仅低级样品,以及ii)来自每个病例的仅高级样品。对于每个视角,流水线计算x倍的平均接受者-操作者曲线下面积(auc),并使用0.5的操作点(在我们的应用中,如果样品评分≥0.5,则患者被预测为gg1,否则为gg≥2)计算x倍中所有患者的真阳性(tp)、假阳性(fp)和真阴性(tn)率。此外,对于这些统计数据中的每一个,流水线都会从两个角度报告值的平均值[例如auc
平均
=(auc
低
+auc
高
)/2]。最后,流水线通过计算交叉验证重复(例如,跨越1000个分区)的中位数统计数据来进一步总结。
[0135]
分级组分类器pronto-e和pronto-m的验证
[0136]
我们运行所述流水线以详尽地测试其支持的所有可能的方法,从而使得能够更彻底地检索最佳方法。选择验证方法有两个主要因素。首先,我们想要从交叉验证产生更大auc值的方法,因为它们表明相应分类器的总体性能更好。其次,我们赞成更高的tp率(即tp率≥0.8),因为这优先于对gg≥2病例的正确分类,这与我们与临床医师的商讨一致,临床医师优先对这些病例进行早期干预,但代价为过度治疗一些gg1病例(由fp率量化)。25个表现最好的分类器的auc为0.772至0.790(图2),并且其中大多数使用正则化判别分析或支持向量机。pronto-m是唯一满足tp率约束(tp率=0.800,auc=0.774)的前25个分类器,并且
我们还选择了pronto-e(tp率=0.833,auc=0.770)进行验证。表5描述了用于产生这两个分类器的方法。
[0137]
然后使用每种选定的方法来训练具有未分区训练组群的分类器,限于具有所需样品和特征数据的患者。与交叉验证一样,我们计算了平均auc、tp和fp率,其中平均值是针对仅低级样品的值和针对仅高级样品的值。尽管已知瘤内异质性s[53],但在诊断时,尚不清楚活检样品的分级在多大程度上代表整个肿瘤的总体分级。为了更好地模拟这种临床情况,对于验证组群中的每位患者,随机选择一个样品,使用代表性样品来计算统计数据,并将此过程重复1000次。我们计算了这些重复的中位auc、tp和fp率(即基于采样的统计数据)。
[0138]
分子谱之间的相似性
[0139]
在该分析中,我们计算了来自同一患者的样品的分子谱之间的相似性(即低级样品谱与高级样品谱之间的相似性),因此,只有一个样品的患者被排除在外。对于所有平台,我们只考虑没有缺失值的谱(对于任何特征)。对于can谱,该谱首先被限制为来自mlpa平台的特征,因为经验证的分类器仅使用来自该平台的cna特征。我们将cna谱之间的成对相似性定义为其中两个样品具有相同cna状态(即改变或未改变)的特征部分。对于rna丰度和甲基化谱,我们将成对相似性定义为特征值之间的一致性系数。
[0140]
单变量特征分析
[0141]
对于每个平台,我们分别测试了每种特征与病理gg的单变量关联(即gg1与gg≥2)。具体来说,我们随机选择了每个病例的一个样品,并随后对于每种特征,使用所选样品来量化gg≥2与gg1病例的特征值差异,x(gg≥2)-x(gg1),并估计所述差异的显著性。对于rna和甲基化平台,我们将x(gg1)和x(gg≥2)分别定义为gg1和gg≥2病例的中位数特征值,并使用双侧曼-惠特尼检验比较gg1和gg≥2病例的特征值集来评价显著性。对于cna平台,我们将x(gg1)和x(gg≥2)分别定义为具有鉴定的cna的gg1和gg≥2病例的比例,并使用双侧比例检验来估计显著性。在来自同一平台的所有特征中,使用benjamini-hochberg方法来调整来自统计测试的p值(产生q值)。将采样程序和随后的统计学计算重复1000次,允许计算重复的中位数、第一和第三四分位数值。该特征分析在训练和验证数据下分开进行。为了估计两个组群中给定特征的单变量关联的显著性,我们使用加权z方法来组合每个组群的中位数q值,通过用于计算它的病例数对每个q值加权s[54].
[0142]
结果
[0143]
组群/样品概述
[0144]
我们成功地为来自训练和验证组群的535个前列腺切除术病例的样品产生了954个mrna、845个nanostring-cna、794个mlpa-cna和847个甲基化谱。我们还为492个病例产生了capra评分。
[0145]
gg分类器的开发和验证
[0146]
分类器针对来自两个位点的333个病例进行了训练,保留了来自第三个位点的202个病例以进行独立验证(表4)。在我们评价的>41,000个gg分类器中,718个表现出auc≥0.75而tp和tn率≥0.5(即每个gg类中≥50%的病例被正确预测)。由于临床需要早期干预,因此相对于特异性优先化对gg≥2的敏感性,因此我们选择了两个性能最佳的分类器进行验证,即pronto-e和pronto-m(表5)。对于具有gg>2样品的病例,这两个分类器都使用来自
m二者都代表了对当前方法的显著改进。将三种商业上可获得的生物标志物测试设计用于活检组织,以在诊断时为早期cap的管理提供信息[40]。prolaris使用与临床/病理参数组合的细胞周期进展基因的rna表达数据(myriad genetics),并报告了10年前列腺特异性死亡率的风险[41]。鉴于cap通常在50至65岁时被诊断出来,并且绝大多数死亡发生在诊断之后20至25年[42],prolaris可能不适合围绕as做出决策。oncotypedx前列腺(genomic health)(基于17种基因qpcr的测试)和promark(metamark genetics)(原位定量蛋白质组测试[22,43])二者都使用活检样品来预测不良病理状况,定义为gg≥3和/或癌症扩散到前列腺外[19,20,22]。值得注意的是,目前可用的测试都没有准确地对gg1与gg≥2病例进行分类,这使得这些中风险患者处于选择as的灰色区。与单独的capra(auc=0.63)相比,将oncotypedx基因组前列腺评分(gps)添加至capra临床和病理列线图非常轻微地提高了不良病理状况的auc(auc=0.67)[20,44]。promark的表现稍好一些,独立的“有利病理”使在活检时产生的auc为0.69[19],当仅用于nccn(national comprehensive cancer network,国家综合癌症网络)指南分类为有利风险的患者时,auc提高至0.75[2,45]。
[0158]
尽管检测gg≥2的活检准确度有限,但as的金标准终点,oncotypedx和promark二者均报道了对肿瘤异质性的抗性[19,20]。这些结果表明,存在可测量的潜在克隆变化,其介导了cap侵袭性,反映了整个肿瘤的gg,并且始终存在于表型肿瘤异质性的区域[46,47]。目前的工作导出并独立地验证了两种新gg分类器,其显示出对肿瘤异质性的抗性,产生了基于采样的auc为0.799(pronto-e)和0.786(pronto-m)。两种分类器都可以使用表型低级肿瘤样品的分子特征来检测gg≥2肿瘤(图3e)。
[0159]
pronto-e包含353种特征,分为mrna丰度和dna cna类型。更紧凑的parse-m包含94种特征,分为mrna丰度、dna cna和dna甲基化类型,并且包括术前临床和病理特征(年龄、临床阶段和psa、活检gg)。虽然来源于前列腺切除术组织(对于其gg最准确),但两种分类器都对采样误差具有抗性,并因此很大概率是当用于活检组织时,它们将更好地为围绕as与临床管理的决策提供信息。目前正在进行这样的工作:使用来自具有统计能力的组群的活检样品来验证分类器。
[0160]
当对同一患者进行时,oncotypedx和prolaris经常产生相互矛盾的建议[48]。尽管如此,这些测试已经表明减少活检频率和过度治疗的潜能[40]表明更准确的测试具有相似的(如果不是更好的话)潜在影响。一旦pronto-e和pronto-m的性能在核心活检中得到验证,这些测定就有可能显著改善这种影响。将每个经验证分类器应用于来自1000名被选为as的假设男性的诊断活检的建模相对简单,假设这些男性中的33%在其as期间会升级[49]。一项性能特征类似于pronto-m的检验将53.4%的男性鉴定为阳性(对于隐匿性gg≥2的风险)以及将46.6%鉴定为阴性。在那些检验呈阳性的人(534/1000男性)中,267人将是tp,并受益于早期重复活检和治疗。在466名检验呈阴性的男性中,只有13.5%(63名)将是假阴性。对于具有fp结果的所有病例中的26.7%,我们建议结果是更早的第一次as活检,而不是另外的活检。这些患者的早期活检将为低gg疾病提供病理保证,而不增加发病率。pronto-e的假设结果相似(图9)。随着时间推移,这样的检验的使用可能会降低对大部分被鉴定为风险较低的患者的监测,并在群体基础上减少进行的活检程序的数目。
[0161]
目前的工作将pronto-e和pronto-m确立为gg的分子生物标志物,它们对采样误差具有抗性,并因此可在诊断活检中表现良好。需要并正在进行进一步的工作,以充分验证其
临床性能。多灶性cap代表了任何活检测试的潜在缺陷,因为活检可能会采样次要的低级病灶,而无法采样更高级的“主要”或“指标(index)”病灶。据估计,这种现象解释了20至30%在活检与前列腺切除术之间升级的病例[15,50]。分类器对活检组织的性能也可能因限制小活检组织样品的核酸产量而受到影响。这种限制应通过预期提高相对于手术样品的活检中分类器性能的因素来平衡,包括在活检组织中观察到的更高品质的核酸[51]以及在临床测定中采用更灵敏和精确的大规模平行测序技术[52]的机会。
[0162]
虽然数项研究将活检分类器与手术之后的结果联系起来,但很少有信息将测试结果与男性as的结果联系起来。需要进一步验证在来自as男性的活检上的pronto-e和pronto-m。总体而言,这些结果表明,将转录组、表观基因组和基因组特征组合可以提高临床相关生物标志物对cap组织的性能。该结果表明对其他生物样本类型(例如血液或尿)和肿瘤位点的潜在益处。
[0163]
尽管本文中已经描述了本发明的优选实施方案,但是本领域技术人员将理解,在不脱离本发明的精神或所附权利要求书的范围的情况下可以对其进行变化。本文中公开的所有文件,包括以下参考文献列表中的那些,均通过引用并入。
[0164]
参考文献列表
[0165]
1.esserman l,shieh y,thompson i.rethinking screening for breast cancer and prostate cancer.jama 2009;302(15):1685-92.
[0166]
2.mohler j,bahnson rr,boston b,et al.nccn clinical practice guidelines in oncology:prostate cancer.j natl compr canc netw 2010;8(2):162-200.
[0167]
3.humphrey pa,moch h,cubilla al,et al.the 2016 who classification of tumours of the urinary system and male genital organs-part b:prostate and bladder tumours.eur urol 2016;70(1):106-119.
[0168]
4.eggener se,scardino pt,walsh pc,et al.predicting 15-year prostate cancer specific mortality after radical prostatectomy.j urol 2011;185(3):869-75.
[0169]
5.ross hm,kryvenko on,cowan je,et al.do adenocarcinomas of the prostate with gleason score(gs)</=6 have the potential to metastasize to lymph nodes?am j surg pathol 2012;36(9):1346-52.
[0170]
6.kelly rs,sinnott ja,rider jr,et al.the role of tumor metabolism as a driver of prostate cancer progression and lethal disease:results from a nested case-control study.cancer metab 2016;4:22.
[0171]
7.cutruzzola f,giardina g,marani m,et al.glucose metabolism in the progression of prostate cancer.front physiol2017;8:97.
[0172]
8.levesque c,nelson ps.cellular constituents of the prostate stroma:key contributors to prostate cancer progression and therapy resistance.cold spring harb perspect med 2018;8(8).
[0173]
9.beltran h,hruszkewycz a,scher hi,et al.the role of lineage plasticity in prostate cancer therapy resistance.clin cancer res 2019;25(23):
prostate tumors.eur urol 2017;72(1):22-31.
[0253]
s35.fraser m,sabelnykova vy,yamaguchi tn,et al.genomic hallmarks of localized,non-indolent prostate cancer.nature 2017;541(7637):359-364.
[0254]
s36.taylor bs,schultz n,hieronymus h,et al.integrative genomic profiling of human prostate cancer.cancer cell 2010;18(1):11-22.
[0255]
s37.nouri m,caradec j,lubik aa,et al.therapy-induced developmental reprogramming of prostate cancer cells and acquired therapy resistance.oncotarget 2017;8(12):18949-18967.
[0256]
s38.harma v,virtanen j,makela r,et al.a comprehensive panel of three-dimensional models for studies of prostate cancer growth,invasion and drug responses.plos one 2010;5(5):e10431.
[0257]
s39.warde-farley d,donaldson sl,comes o,et al.the genemania prediction server:biological network integration for gene prioritization and predicting gene function.nucleic acids res 2010;38(web server issue):w214-20.
[0258]
s40.franceschini a,szklarczyk d,frankild s,et al.string v9.1:protein-protein interaction networks,with increased coverage and integration.nucleic acids res 2013;41(database issue):d808-15.
[0259]
s41.orr b,riddick ac,stewart gd,et al.ldentification of stromally expressed molecules in the prostate by tag-profiling of cancer-associated fibroblasts,normal fibroblasts and fetal prostate,oncogene2012;31(9):1130-42.
[0260]
s42.vanpoucke g,orr b,grace oc,et al.transcriptional profiling of inductive mesenchyme to identify molecules involved in prostate development and disease.genome biol 2007;8(10):r213.
[0261]
s43.boufaied n,nash c,rochette a,et al.identification of genes expressed in a mesenchymal subset regulating prostate organogenesis using tissue and single cell transcriptomics,sci rep 2017;7(1):16385.
[0262]
s44.sun y,goodison s.optimizing molecular signatures for predicting prostate cancer recurrence.prostate 2009;69(10):1119-27.
[0263]
s45.sboner a,demichelis f,calza s,et al,molecular sampling of prostate cancer:a dilemma for predicting disease progression.bmc med genomics 2010;3:8.
[0264]
s46.mortensen mm,hoyer s,lynnerup as,et al.expression profiling of prostate cancer tissue delineates genes associated with recurrence after prostatectomy.sci rep 2015;5:16018.
[0265]
s47.roberto d,selvarajah s,park pc,et al.functional validation of metabolic genes that distinguish gleason 3 from gleason 4 prostate cancer foci.prostate 2019;79(15):1777-1788.
[0266]
s48.patel pg,wessel t,kawashima a,et al.a three-gene dna methylation biomarker accurately classifies early stage prostate cancer.prostate 2019;79
(14):1705-1714.
[0267]
s49.cooperberg mr,broering jm,carroll pr.risk assessment for prostate cancer metastasis and mortality at the time of diagnosis.j natl cancer inst 2009;101(12):878-87.
[0268]
s50.waggott d,chu k,yin s,et al.nanostringnorm:an extensible r package for the pre-processing of nanostring mrna and mirna data.bioinformatics 2012;28(11):1546-8.
[0269]
s51.d
′
ambrosio a,mazzeo g,lorio c,et al.a differential evolution algorithm for finding the median ranking under the kemeny axiomatic approach.computers&operations research 2017;82:126-138.
[0270]
s52.bischl b,lang m,kotthoff l,et al.mlr:machine learning in r,journal of machine learning 2016;17:1-5.
[0271]
s53.boutros pc,fraser m,harding nj,et al.spatial genomic heterogeneity within localized,multifocal prostate cancer.nat genet 201547(7):736-45.
[0272]
s54.whitlock mc.combining probability from independent tests:the weighted z-method is superior to fisher
′
s approach.j evol biol 2005;18(5):1368-73.
[0273]
表1.训练组群和验证的临床病理特征。
[0274]
[0275]
[0276][0277]
*使用反向kaplan-meier方法计算[53]
[0278]
表2.分类器性能
[0279][0280]a平均值代表从来源于低级和高级样品的值计算的平均值。
[0281]b基于采样的统计数据提供了对临床实践(见方法)的更好地代表。缩写:tpr-真阳性率;fpr-假阳性率;tnr-真阴性率;fnr-假阴性率;auc-(接受者-操作者)曲线下面积。
[0282]
表3.当地伦理批准列表。
[0283][0284]
*收集医院的样品在来自多伦多大学伦理委员会的批准下在安大略癌症研究所(ontario institute for cancer research,oicr)进行处理,该委员会是oicr的研究伦理记录委员会。
[0285]
表4.训练组群和验证组群的consort表。
[0286][0287]a对于训练组群中的146个病例和验证组群中的25个病例,主要核心活检材料无法用于中央病理审查,导致这些病例被排除在外。
[0288]b病理审查之后排除了另外34和16个病例(gg>2等)。
[0289]c分别有34个和5个病例没有获取/获取不完整的分子数据。
[0290]
表5.用于产生分类器的方法
[0291][0292][0293]
表6.gg分类器的候选特征。
[0294]
特征表
[0295]
gg分类器的候选特征列表、相应的单变量分析结果(见补充方法)以及经验证gg分类器中的成员资格指标。对于二元特征,训练和验证差异被定义为比例差异。
[0296][0297]
cna特征比较表
[0298]
来自mlpa和nanostring测定的cna特征的比较。对于nanostring测定,将1至3个基因折叠成特征性特征(因此,nanostring特征可能与多个基因行相关)。
[0299][0300]
[0301]
[0302]
[0303]
[0304]
[0305]
[0306]
[0307]
[0308]
[0309]
[0310]
[0311]
[0312][0313]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1