一种二硫醚类化合物及其用于催化烷基芳香烃化合物苄位氧化的方法
1.本发明属于有机化工合成技术领域,具体涉及一种二硫醚类光催化剂及一种烷基芳香烃类化合物苄位氧化制备酮的方法。
技术背景
2.由于有机二硫化物具有很强的电子转移和产生自由基能力,在可见光催化分子氧氧化领域中炙手可热:2008年,tsuboi课题组报道了在室温下由二苯二硫衍生物催化的烯丙醇向丙烯醛的光诱导氧化(bull.chem.soc.jpn.2008,81,361-368.);2016年,王磊课题组报道了可见光激发形成的巯基自由基用于催化分子氧氧化内炔类化合物制备1,2-二酮类化合物(green chem.2016,18,6373-6379.);2017年王晓课题组通过实验与计算的结合,利用富电子芳香二硫醚作为催化剂,氧气作为氧化剂,在可见光条件下实现了单、多取代芳香族烯烃在常温下向醛或酮的转化(angew.chem.,int.ed.2017,56,832-836.);2019年,孟庆伟课题组报道了二硫化物作为光催化剂分别实现酮酸酸羟基化及羟甲基化反应(chemical communications,2019,55:13008-13011;108558665 a;108752213 a)。综上所述,二硫化物介导可见光催化的分子氧氧化反应仅在烯烃、炔烃和烯丙醇的氧化少量报道,而对sp
3 c-h键的分子氧氧化反应尚未见公开报道,这是由于sp
3 c-h键相较于sp2和sp c-h键,具有更高的键能难以活化。
3.c-h键活化是构建复杂有机分子的有力工具之一,sp和sp
2 c-h键的活化通常需要过渡金属进行催化,而自由基c-h键活化在sp
3 c-h键的活化中显示出巨大的应用价值。要实现自由基c-h键活化应选择合适的自由基引发体系。通常,重氮化合物或过氧化物在高温或光引发条件下能够诱导c-h键生成自由基。目前最常用的自由基引发剂就是过氧化物(如tbhp\dtbp等),因为过氧化物会发生均裂产生烷氧自由基,通常需要高温或者在反应物中加入低价金属来促进烷氧自由基产生,从而从反应底物中提取h产生自由基中间体,最终实现c-h键自由基活化。但是过氧化物性质不稳定而且容易爆炸,大量使用过氧化物对工业生产上具有限制,高温及金属催化剂的使用会造成大量能源的浪费。因此亟需开发一种新的技术,在温和条件下实现一系列苄位sp
3 c-h键氧化。
技术实现要素:
4.针对现有技术存在的技术问题,本发明使用廉价易得且结构简单的二硫醚化合物作为一种新型自由基引发剂,无外加金属,碱,自由基引发剂和还原剂,在可见光的作用下,实现底物苄位sp
3 c-h键活化并进行一系列的需氧氧化反应。
5.本发明的第一方面在于,提供一类用于催化烷基芳香烃化合物苄位氧化的二硫醚类化合物;所述的二硫醚类化合物如式i所示:
6.7.其中,r1和r2分别独立地选自苯基、五元芳杂环或六元芳杂环中的一种;
8.对于上文所述的技术方案中,进一步优选的,所述r1,r2为苯环基时,如下式:
[0009][0010]
其中,每个苯环上具有相邻于、相间于或对位于二硫键的取代基r9、r
10
;且所述r9和r
10
分别独立地选自卤素、no2、氰基、c1-c8烷基或c1-c8烷氧基中的一种。
[0011]
对于上文所述的技术方案中,进一步优选的,所述r1、r2分别独立地选自吡啶基、嘧啶基、噻吩基、苯并噻唑基中的一种;
[0012]
对于上文所述的技术方案中,进一步优选的,所述的二硫醚化合物(式i)为苯并噻唑二硫化物时,其结构式如式ia:
[0013][0014]
本发明的第二方面在于,公开一种可见光激发二硫醚类化合物催化烷基芳香烃化合物苄位氧化的方法,该方法可制备芳香酮或芳香醛;其包括如下步骤:在溶剂中,以结构式如i所示的二硫醚化合物作光敏催化剂,分子氧作氧化剂,可见光作为激发光源,使烷基芳香烃类化合物的苄位c-h键直接氧化生成芳香酮类化合物,或使芳香苄胺类化合物氧化成醛。
[0015]
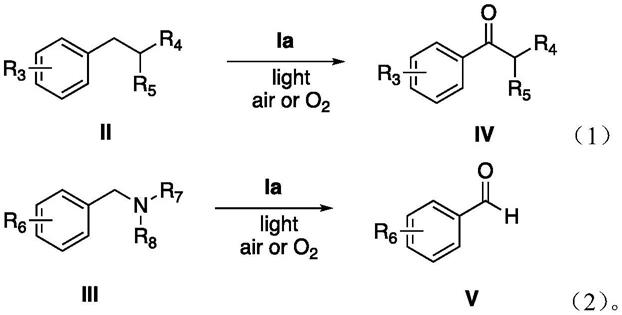
对于上文所述的技术方案中,进一步优选的,所述的烷基芳香烃类化合物如式ii所示:
[0016][0017]
其中,r3选自氢原子、卤素、烷基、烷氧基中的一种;r4和r5分别独立地选自氢原子、烷基、酰氯中的一种;
[0018]
对于上文所述的技术方案中,进一步优选的,所述的芳香苄胺类化合物如式iii所示:
[0019][0020]
其中,r6选自氢原子、卤素、烷基、烷氧基中的一种;r7和r8分别独立地选自氢原子、烷基、苯甲酰基、酰氯中的一种;
[0021]
对于上文所述的技术方案中,进一步优选的,所述的二硫醚化合物(式i)为苯并噻唑二硫化物时,对应的光催化反应式如式(1)及(2):
[0022][0023]
对于上文所述的技术方案中,进一步优选的,所述光催化反应的温度为-30~100℃;结构式如i所示的二硫醚类化合物与底物的摩尔比是0.001~0.5:1,所述可见光的波长为365nm-700nm。
[0024]
对于上文所述的技术方案中,进一步优选的,所述的光催化反应的温度为10℃~50℃;所述二硫醚类化合物与底物的摩尔比为0.005~0.5:1。
[0025]
对于上文所述的技术方案中,进一步优选的,所述的溶剂选自水、甲苯、对二甲苯、邻二甲苯、均三甲苯、正己烷、四氢呋喃、乙酸乙酯、乙腈、dmf、dmap、dmso、氯仿、四氯化碳、二氯甲烷、溴甲烷、二溴甲烷、1,2-二氯乙烷、1,3-二溴丙烷、二硫化碳、二氧六环、石油醚、甲醇、乙醇、四氢萘、吗啉中的至少一种。
[0026]
对于上文所述的技术方案中,进一步优选的,所述的分子氧是纯氧气、空气或含有氧分子混合物。
[0027]
本发明的有益效果:本发明使用商业易得、廉价稳定的二硫醚类化合物作为催化剂,在可见光照射下实现苄位c-h键氧化。反应条件温和,不需要外加金属、碱、还原剂等即可有效制备羰基和醛基化合物,操作简单,具有良好的底物适用性以及环境友好性,成本低,适合工业规模生产。
具体实施方式
[0028]
下面结合技术方案对本发明的具体实施方式进行详细地描述,但应当理解本发明的保护范围并不受具体实施方式的限制。
[0029]
实施例1制备1-四氢萘酮
[0030]
向2ml乙腈溶液中加入四氢萘(66.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到1-四氢萘酮64.3mg,收率88%。
[0031]1h nmr(400mhz,chloroform-d)δ8.03(dd,j=7.8,1.4hz,1h),7.46(td,j=7.8,1.4hz,1h),7.36
–
7.10(m,2h),2.96(t,j=6.1hz,2h),2.65(t,j=7.3,6.1hz,2h),2.27
–
1.97(m,2h).
[0032]
实施例2制备1-茚酮
[0033]
向2ml乙腈溶液中加入茚满(59.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到1-茚酮49.5mg,收率75%。
[0034]
1h nmr(400mhz,chloroform-d)δ8.29
–
6.82(m,4h),4.99
–
2.91(m,2h),2.62(q,j=8.8,5.4hz,2h).
[0035]
实施例3制备9-芴酮
[0036]
向2ml乙腈溶液中加入芴(83.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到9-芴酮81.1mg,收率90%。
[0037]1h nmr(400mhz,chloroform-d)δ7.57(d,j=7.3hz,2h),7.47
–
7.33(m,4h),7.20(td,j=7.3,1.5hz,2h).
[0038]
实施例4制备二苯基甲酮
[0039]
向2ml乙腈溶液中加入二苯基甲烷(84.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到二苯基甲酮84.7mg,收率93%。
[0040]
实施例5制备苯乙酮
[0041]
向2ml乙腈溶液中加入乙苯(53.08mg,0.5mmol)和苯并噻唑二硫醚(33.2mg,0.1mmol),在氧气氛围下紫光照射36小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到苯乙酮43.8mg,收率73%。
[0042]
1h nmr(400mhz,chloroform-d)δ7.99
–
7.88(m,2h),7.58
–
7.49(m,1h),7.44(tt,j=6.7,1.4hz,2h),2.58(s,3h).
[0043]
实施例6制备对甲氧基苯乙酮
[0044]
向2ml乙腈溶液中加入对甲氧基乙苯(68.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到对甲氧基苯乙酮66.1mg,收率88%。
[0045]1h nmr(400mhz,chloroform-d)δ7.94(d,j=8.9hz,2h),6.94(d,j=8.9hz,2h),3.87(s,3h),2.56(s,3h).
[0046]
实施例7制备对溴苯乙酮
[0047]
向2ml乙腈溶液中加入对溴乙苯(92.53mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到对溴苯乙酮67.7mg,收率68%。
[0048]1h nmr(400mhz,chloroform-d)δ7.81(d,j=8.5hz,2h),7.59(d,j=8.5hz,2h),2.58(s,3h).
[0049]
实施例8制备邻溴苯乙酮
[0050]
向2ml乙腈溶液中加入邻溴乙苯(92.5mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到邻溴苯乙酮40.8mg,收率41%。
[0051]
1h nmr(400mhz,chloroform-d)δ7.61(dd,j=8.0,1.3hz,1h),7.46(dd,j=7.6,1.8hz,1h),7.37(td,j=7.6,1.3hz,1h),2.63(s,3h).
[0052]
实施例9制备2-甲基-1-苯基丙酮
[0053]
向2ml乙腈溶液中加入异丁基苯(67.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分
离,洗脱剂为石油醚/乙酸乙酯,得到2-甲基-1-苯基丙酮25.2mg,收率34%。
[0054]1h nmr(400mhz,chloroform-d)δ8.02
–
7.92(m,2h),7.62
–
7.51(m,1h),7.46(dd,j=8.2,6.8hz,2h),3.57(h,j=6.8hz,1h),1.22(d,j=6.8hz,6h).
[0055]
实施例10制备苯己酮
[0056]
向2ml乙腈溶液中加入苯己烷(81.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到苯己酮49.3mg,收率56%。
[0057]1h nmr(400mhz,chloroform-d)δ8.43
–
7.92(m,2h),7.61
–
7.50(m,1h),7.46(t,j=7.6hz,2h),2.96(t,j=7.4hz,2h),1.74(dd,j=9.1,5.7hz,2h),1.37(q,j=3.8hz,4h),1.13
–
0.77(m,3h).
[0058]
实施例11制备2-氯乙酰基苯
[0059]
向2ml乙腈溶液中加入2-(氯乙基)苯(70.3mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到2-氯乙酰基苯27.0mg,收率35%。
[0060]1h nmr(400mhz,chloroform-d)δ7.89(d,j=7.6hz,2h),7.58
–
7.52(m,1h),7.43(t,j=7.6hz,2h),4.65(s,2h).
[0061]
实施例12制备苯甲酸甲酯
[0062]
向2ml乙腈溶液中加入苄甲醚(61.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到苯甲酸甲酯31.3mg,收率46%。
[0063]1h nmr(400mhz,chloroform-d)δ8.13
–
7.92(m,2h),7.68
–
7.49(m,1h),7.50
–
7.35(m,2h),3.91(s,3h).
[0064]
实施例13制备2-正丁酰噻吩
[0065]
向2ml乙腈溶液中加入2-正丁基噻吩(70.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到2-正丁酰噻吩32.4mg,收率42%。
[0066]1h nmr(400mhz,chloroform-d)δ7.85
–
6.83(m,1h),6.59(t,j=7.3hz,1h),6.45(d,j=7.3hz,1h),3.68
–
3.08(m,2h),2.75(t,j=6.4hz,2h),1.92(p,j=6.4hz,2h).
[0067]
实施例14制备2-乙酰基吡啶
[0068]
向2ml乙腈溶液中加入2-乙基吡啶(53.8mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到2-乙酰基吡啶8.5mg,收率14%。
[0069]1h nmr(400mhz,cdcl3)δ8.62(d,j=4.0hz,1h),7.98(d,j=8.0,8.0hz,1h),7.81-7.74(m,1h),7.44-7.39(m,1h),2.65(s,3h).
[0070]
实施例15制备2-乙酰基蒽醌
[0071]
向2ml乙腈溶液中加入2-乙基蒽醌(118.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到2-乙酰基蒽醌111.4mg,收率89%。
[0072]1h nmr(400mhz,chloroform-d)δ8.74(d,j=1.6hz,1h),8.56
–
8.13(m,4h),7.82
(dd,j=5.9,3.3hz,2h),2.74(s,3h).
[0073]
实施例16制备4-乙酰基联苯
[0074]
向2ml乙腈溶液中加入4-乙基联苯(91.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到4-乙酰基联苯88.3mg,收率90%。
[0075]1h nmr(400mhz,chloroform-d)δ8.09
–
7.99(m,2h),7.77
–
7.65(m,2h),7.66
–
7.60(m,2h),7.55
–
7.44(m,2h),7.43
–
7.36(m,1h),2.64(s,3h).
[0076]
实施例17制备环己基苯基甲酮
[0077]
向2ml乙腈溶液中加入(环己基甲基)苯(87.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到环己基苯基甲酮69.6mg,收率74%。
[0078]
1h nmr(400mhz,chloroform-d)δ8.02
–
7.86(m,2h),7.57
–
7.49(m,1h),7.45(dd,j=8.3,6.7hz,2h),3.26(tt,j=11.5,3.3hz,1h),1.98
–
1.76(m,4h),1.73(dtt,j=12.7,3.4,1.6hz,1h),1.57
–
1.13(m,5h).
[0079]
实施例18制备二苯基乙酮
[0080]
向2ml乙腈溶液中加入1,2-二苯乙烷(91.1mg,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),在氧气氛围下紫光照射16小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到二苯基乙酮88.3mg,收率61%。
[0081]1h nmr(400mhz,chloroform-d)δ8.10
–
7.97(m,2h),7.77
–
7.66(m,2h),7.65
–
7.60(m,2h),7.54
–
7.44(m,2h),7.43
–
7.34(m,1h),2.64(d,j=1.0hz,3h).
[0082]
实施例19制备苯甲醛(式iii,其中r1为h)
[0083]
向3ml chcl3溶液中加入苄胺(式ii,其中r1为h,0.0536g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,苄胺》99%转化率,生成苯甲醛,收率74%。
[0084]
实施例20制备苯甲醛(式iii,其中r1为h)
[0085]
向3ml chcl3溶液中加入苄胺(式ii,其中r1为h,0.0536g,0.5mmol)和噻吩二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成苯甲醛,收率60%。
[0086]
实施例21制备对甲基苯甲醛(式iii,其中r1为4-me)
[0087]
向3ml chcl3溶液中加入对甲基苄胺(式ii,其中r1为4-me,0.0605g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成对甲基苯甲醛,收率72%。
[0088]
实施例22制备3-甲基苯甲醛(式iii,其中r1为3-me)
[0089]
向3ml chcl3溶液中加入3-甲基苄胺(式ii,其中r1为3-me,0.0605g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成3-甲基苯甲醛,核磁收率51%。
[0090]
实施例23制备4-甲氧基苯甲醛(式iii,其中r1为4-ome)
[0091]
向3ml chcl3溶液中加入4-甲氧基苄胺(式ii,其中r1为4-ome,0.0605g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成4-甲氧基苯甲醛,核磁收率77%。
[0092]
实施例24制备2-甲氧基苯甲醛(式iii,其中r1为2-ome)
[0093]
向3ml chcl3溶液中加入2-甲氧基苄胺(式ii,其中r1为2-ome,0.0605g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成2-甲氧基苯甲醛,核磁收
率61%。
[0094]
实施例25制备4-叔丁基苯甲醛(式iii,其中r1为4-t-bu)
[0095]
向3ml chcl3溶液中加入4-叔丁基苄胺(式ii,其中r1为4-t-bu,0.0816g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成4-叔丁基苯甲醛,核磁收率83%。
[0096]
实施例26制备2-氟苯甲醛(式iii,其中r1为2-f)
[0097]
向3ml chcl3溶液中加入2-氟苄胺(式ii,其中r1为2-f,0.0626g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成2-氟苯甲醛,收率68%。
[0098]
实施例27制备苯甲醛(式iii,其中r1为h)
[0099]
向3ml chcl3溶液中加入n-甲基苄胺(式ii,其中r1,r3为h,其中r2为me,0.0606g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成苯甲醛,核磁收率62%。
[0100]
实施例28制备苯甲醛
[0101]
向3ml chcl3溶液中加入n,n-二甲基苄胺(式iii,其中r6为h,r7,r8为me,0.0726g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成苯甲醛,收率49%。
[0102]
实施例29制备苯甲醛
[0103]
向3ml chcl3溶液中加入苄胺(式iii,其中r6,r8为h,其中r7为i-pr,0.0746g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成苯甲醛,核磁收率43%。
[0104]
实施例30
[0105]
向3ml chcl3溶液中加入苄胺(式iii,其中r6,r8为h,其中r7为ph,0.1556g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成苯甲醛,核磁收率44%。
[0106]
实施例31
[0107]
向3ml chcl3溶液中加入n-苯甲酰基苄胺(式iii,其中r6,r8为h,其中r7为苯甲酰基,0.0916g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成苯甲醛,核磁收率70%;生成苯甲酰胺,核磁收率81%。
[0108]
实施例32
[0109]
向3ml chcl3溶液中加入n-苯甲酰基对甲氧基苄胺(式iii,其中r6为4-ome,r8为h,其中r7为苯甲酰基,0.1206g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成对甲氧基苯甲醛,核磁收率66%;生成苯甲酰胺,核磁收率68%。
[0110]
实施例33
[0111]
向3ml chcl3溶液中加入n-(对甲氧基)苯甲酰基苄胺(式iii,其中r6为h,r8为h,其中r7为对甲氧基苯甲酰基,0.1206g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成对甲氧基苯甲醛,核磁收率83%;生成对甲氧基苯甲酰胺,核磁收率77%。
[0112]
实施例34
[0113]
向3ml chcl3溶液中加入n-甲酰氯苄胺(式iii其中r6为,r8为h,其中r7为甲酰氯,
0.0918g,0.5mmol)和苯并噻唑二硫醚(16.6mg,0.05mmol),10w紫光照射8小时,生成苯甲醛,核磁收率42%。
[0114]
实施例35制备苯甲醛
[0115]
向3ml chcl3溶液中加入苄胺(式iii,其中r6为h,0.0536g,0.5mmol)和苯并噻唑二硫醚(0.0830g,0.25mmol),10w紫光照射8小时,生成苯甲醛,收率80%。
[0116]
对比例1制备苯乙酮
[0117]
向2ml乙腈溶液中加入乙苯(53.08mg,0.5mmol)和二苯二硫醚(21.8mg,0.1mmol),在氧气氛围下紫光照射36小时。几乎没有得到产物。
[0118]
对比例2制备苯乙酮
[0119]
向2ml乙腈溶液中加入乙苯(53.08mg,0.5mmol)和4-氟二苯二硫醚(21.8mg,0.1mmol),在氧气氛围下紫光照射36小时。反应完成后,减压浓缩除去溶剂乙腈,柱层析分离,洗脱剂为石油醚/乙酸乙酯,得到苯乙酮4.2mg,收率仅为7%。
[0120]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,本领域的技术人员在本发明披露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求书的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1