一种泛影酸的合成方法与流程
1.本发明属于化学制药技术领域,涉及一种泛影酸的合成方法。
背景技术:
2.泛影酸,化学名:3,5-二乙酰氨基-2,4,6-三碘苯甲酸,结构式:
[0003][0004]
泛影酸作为x线造影剂,为x线诊断用阳性造影剂,供配制泛影葡胺、泛影酸钠或复方泛影葡胺注射液后应用。泛影酸在医学造影方面的应用非常广泛。其作为一种常用的ct造影剂,由于其存在两个氨基活性基团,且由于在ich指导原则中,芳香胺作为基因毒性警示结构需要重点控制,杂质限度较低,所以该产品的杂质含量作为一个技术壁垒导致国内外该产品的制备公司较少,可供查阅的文献资料并不全面,其中主要包含以下几个工艺路线。
[0005]
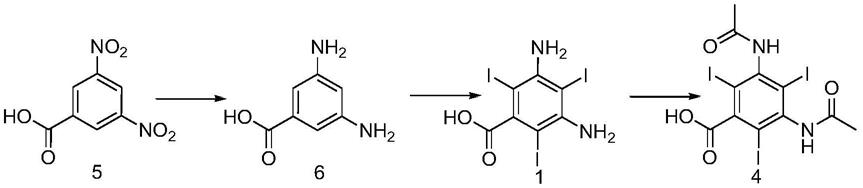
在journal of the american chemical society,1956,vol.78,p.3210,3215中记载的以3,5-二硝基苯甲酸(化合物5)作为原料,还原得到3,5-二氨基苯甲酸(化合物6),进一步的碘化制备3,5-二氨基-2,4,6-三碘苯甲酸(化合物1),通过乙酸酐酰化得到3,5-二乙酰氨基苯甲酸(化合物4),得到泛影酸粗品,该文献中对于泛影酸的精制提纯方式基本未进行描述,反应路线为:
[0006]
该路线制备过程中,由于碘化物(3,5-二氨基-2,4,6-三碘苯甲酸)在与乙酸酐进行酰胺化过程中,泛影酸在乙酸及乙酸酐中均无法溶解,导致反应过程中未反应完全的3,5-二氨基-2,4,6-三碘苯甲酸以及3-乙酰氨基-5氨基-2,4,6-三碘苯甲酸会随着产物(泛影酸)的析晶,而包裹在晶体内部一起析出,在后续的精制提纯中较难分离提纯,由于芳香胺作为ich中明确指出的基因毒性杂质,且在各国药典中,对于这类杂质的限度要求均非常高,例如ep10.0药典中对于杂质a(单酰胺化合物)及杂质d控制限度约为0.01%(自身对照浓度),近乎于要求未检出。
[0007][0008]
另外还有文献报道采用羟甲基树脂键合化合物1后,通过传统工艺酰胺化,然后脱去羟甲基树脂的工艺,但该工艺存在成本高,且实际生产过程中,树脂回收率低,收率及产量受限等缺点。
[0009]
而对于泛影酸精制的相关文献大多采用在水中碱溶酸析,以及大量活性炭吸附杂质来达到除杂及脱色效果,使得实际生产过程中,操作繁琐,物料损耗大,收率低,以及产生的三废高等缺点。
[0010]
针对该产品较为严格的杂质限度要求,使得传统工艺中的碱溶酸析结晶工艺,需要多次重复操作来使得产物达到质量要求,以至于在精制过程中会产生大量的三废及能耗,导致生产成本过高的同时,很难提升产品的产能。
技术实现要素:
[0011]
针对现有技术存在的这些问题,本发明提供一种新的泛影酸合成方法,该合成方法能够较为方便的得到符合国内外药典标准的高纯度的目标化合物,并能够在现有的技术水平上,较大幅度的提高产率及降低反应成本。
[0012]
为了实现发明目的,本发明采取如下的技术方案:
[0013]
一种泛影酸的合成方法,包括如下步骤:
[0014]
(1)在催化剂的存在下,3,5-二氨基-2,4,6-三碘苯甲酸化合物1与氯化亚砜以及乙酸混合反应,得到3,5-二乙酰氨基-2,4,6-三碘苯甲酰氯化合物2;
[0015]
(2)3,5-二乙酰氨基-2,4,6-三碘苯甲酰氯化合物2与醇进行酯化反应得到3,5-二乙酰氨基-2,4,6-三碘苯甲酸酯化合物3;
[0016]
(3)3,5-二乙酰氨基-2,4,6-三碘苯甲酸酯化合物3用碱水解,加盐酸析晶,抽滤,烘干得到泛影酸粗品;
[0017]
(4)将泛影酸粗品升温溶解于氨水甲醇溶液中,降温,加活性炭回流搅拌,趁热过滤,滤液加入盐酸调节ph至中性,降温大量固体,抽滤,烘干,得到泛影酸成品;合成路线如下:
[0018][0019]
进一步地,所述步骤(1)的催化剂选自二甲基甲酰胺,4-二甲氨基吡啶,n,n-二甲基苯胺或吡啶;用到的溶剂选自氯化亚砜,氯仿,二氧六环,乙二醇单甲醚,二甲基乙酰胺,
n-甲基吡咯烷酮。
[0020]
进一步地,所述步骤(1)的氯化亚砜、乙酸与化合物1的摩尔比≥3:≥2:1。即最低要求在1摩尔的化合物1的时候,氯化亚砜的量应至少3摩尔,乙酸的量至少2摩尔,且氯化亚砜的量应保持比乙酸多一摩尔以上,以便于化合物1的羧基酰氯化。
[0021]
进一步地,所述步骤(1)中的反应温度为20-80℃,优选为40-70℃,更优选为45-65℃。
[0022]
进一步地,所述步骤(2)中用hplc中控至酰氯化合物不大于0.5%后停止反应,此时析出大量白色固体,降温至20-25℃析晶,抽滤,滤饼用醇淋洗,烘干。
[0023]
进一步地,所述步骤(2)中的醇为甲醇、乙醇、异丙醇,正丙醇,正丁醇或叔丁醇。
[0024]
进一步地,所述步骤(2)中的酯化反应温度优选为50-110℃,更优选为60-80℃。
[0025]
所述步骤(3)中所述的碱选自氢氧化钠、氢氧化钾或氢氧化锂;在ph10-11环境下进行酯基水解。
[0026]
进一步地,所述步骤(3)中用hplc中控至甲酯化合物含量不大于0.5%后,使用浓盐酸调节ph至3-3.5,搅拌升温至90-95℃,加入活性炭,继续搅拌,趁热过滤,滤液降温至60℃后,滴加浓盐酸调节ph至1-1.5,保温搅拌析晶,抽滤,滤饼用去离子水淋洗,烘干。
[0027]
进一步地,所述步骤(4)中氨水甲醇溶液为浓氨水与甲醇的体积比为5:95。
[0028]
本发明主要通过化合物1(3,5-二氨基-2,4,6-三碘苯甲酸)在氯化亚砜的环境下,对羧基进行酰氯化的同时,加入一定量的乙酸,使乙酸与过量的氯化亚砜反应生成的乙酰氯与氨基酰胺化,得到化合物2(3,5-二乙酰胺基-2,4,6-三碘苯甲酰氯),蒸馏掉过量的酰氯化试剂(氯化亚砜),同时利用酰氯化物的反应液溶清,呈均相反应,避免了大量的芳香氨基化合物反应不完全的问题;再加入醇类酯化,得到化合物3(3,5-二乙酰氨基-2,4,6-三碘苯甲酸甲酯)或其他酯类,利用化合物3与其他副产物在低级醇中溶解度的差别,去除较难去除的部分杂质,最后通过碱性水解乙酰氧基后,酸析出泛影酸粗品,将粗品通过氨水-甲醇溶液中溶解后,酸析得到泛影酸成品,该工艺摩尔收率可以达到80%左右,且该工艺仅需要经过一次重结晶过程,大大的降低了重结晶过程使产生的三废,目前文献记载较多的工艺在经过多次重结晶以及活性炭处理后,以化合物1(3,5-二氨基-2,4,6-三碘苯甲酸)计算,摩尔收率仅达到60-65%。
附图说明
[0029]
图1是实施例4中的泛影酸的hplc图谱;
[0030]
图2是实施例7中的泛影酸的hplc图谱.
具体实施方式
[0031]
实施例1:3,5-二乙酰氨基-2,4,6-三碘苯甲酰氯的制备
[0032]
将30g的3,5-二氨基2,4,6-三碘苯甲酸(0.0566mol)加入反应瓶内,加入30g乙酸(0.5mol)升温至50℃溶解,溶清后移入滴液漏斗中备用;将300ml(4.13mol)氯化亚砜加入反应瓶内,加入0.5g的4-二甲氨基吡啶作为催化剂,50℃保温搅拌,将滴液漏斗中的溶液滴加进入反应瓶内,滴加速度控制在内温不超过60℃,滴加完毕后,保温反应24h,然后减压至60℃蒸干反应瓶内溶剂,得到油状物约40g。
[0033]
实施例1中,4-二甲氨基吡啶可用二甲基甲酰胺,n,n-二甲基苯胺或吡啶代替;这里的氯化亚砜也充当了溶剂,如果选用其他溶剂,例如氯仿,二氧六环,乙二醇单甲醚,二甲基乙酰胺或n-甲基吡咯烷酮,则溶剂量在化合物1的摩尔量的3-10倍范围内,而氯化亚砜作为酰氯化试剂则不得低于3,5-二氨基2,4,6-三碘苯甲酸的3倍摩尔量,同时要满足氯化亚砜比乙酸过量一个摩尔量以用于3,5-二氨基2,4,6-三碘苯甲酸中的羧基酰氯化。
[0034]
实施例2:3,5-二乙酰氨基-2,4,6-三碘苯甲酸甲酯的制备
[0035]
将上述制备的40g 3,5-二乙酰氨基-2,4,6-三碘苯甲酰氯油状物,加入反应瓶内,加入300ml甲醇,升温至回流,回流搅拌反应12h,hplc中控至酰氯化合物不大于0.5%后停止反应,此时析出大量白色固体,降温至20-25℃析晶6h,抽滤,滤饼用10ml甲醇淋洗三次,固体烘干,得到3,5-二乙酰氨基-2,4,6-三碘苯甲酸甲酯33g(0.0525mol),摩尔收率92.76%(以化合物1:3,5-二氨基-2,4,6-三碘苯甲酸计算),hplc含量99.1%。
[0036]
实施例2中的甲醇可用乙醇、异丙醇,正丙醇,正丁醇或叔丁醇代替,这样可得到相应的甲酸酯化合物。
[0037]
实施例3:3,5-二乙酰氨基-2,4,6-三碘苯甲酸的制备
[0038]
将上述33g的3,5-二乙酰氨基-2,4,6-三碘苯甲酸甲酯加入反应瓶内,加入200ml的去离子水,搅拌30min后,滴加20%的氢氧化钠水溶液,滴加至反应瓶内完全溶清,调节ph至10-11,维持该ph,50-55℃保温反应2h,hplc中控至甲酯化合物含量不大于0.5%后,使用浓盐酸调节ph至3-3.5,搅拌升温至90-95℃,加入0.15g的活性炭,继续搅拌3h,趁热过滤,滤液降温至60℃后,滴加浓盐酸调节ph至1-1.5,保温搅拌析晶3h,抽滤,滤饼使用10ml去离子水淋洗3次,烘干,得到泛影酸粗品30g(0.0489mol),摩尔收率86.40%(以化合物1:3,5-二氨基-2,4,6-三碘苯甲酸计算),hplc含量99.3%。
[0039]
实施例3中的氢氧化钠可用氢氧化钾或氢氧化锂代替。
[0040]
实施例4:泛影酸的精制
[0041]
将上一步完成的泛影酸粗品30g加入反应瓶内,将5%氨水甲醇溶液180ml加入反应瓶内,升温至回流溶清后,降温至30℃,加入0.15g活性炭,搅拌1h,抽滤,滤液加入盐酸析晶,析晶3h,过滤,滤饼使用10ml无水甲醇淋洗3次,烘干,得到泛影酸精品28g,质量收率93.33%(粗品精制收率),摩尔总收率80.56%,hplc含量99.98%(ep10.0药典方法检测),ep药典方法hplc图谱详见图1。
[0042]
实施例5:3,5-二乙酰氨基-2,4,6-三碘苯甲酸乙酯的制备
[0043]
将实施例1中得到的油状物加入反应瓶内,加入300ml的乙醇,升温至回流搅拌反应12h,此时应析出大量白色固体,降温20-25℃析晶3h,抽滤,滤饼使用10ml的乙醇淋洗3次,烘干,得到白色固体34.3g(0.0534mol),摩尔收率94.39%,hplc含量:99.3%。
[0044]
实施例6:3,5-二乙酰氨基-2,4,6-三碘苯甲酸的制备
[0045]
将实施例5制备的白色固体加入反应瓶内,加入200ml的去离子水,搅拌30min后,滴加液碱调节ph至10-11,升温至50-55℃,保温搅拌反应3h,维持反应液ph在10-11,3h后hplc中控反应液中乙酯化合物不大于0.5%后,浓盐酸调节ph至3-3.5,升温至90-95℃,加入0.15g活性炭,搅拌3h后,趁热过滤;滤液降温至60-65℃后,加入浓盐酸调节ph至1-1.5,保温搅拌析晶3h,抽滤,滤饼使用10ml的去离子水淋洗两次后,烘干,得到泛影酸粗品30.4g(0.0495mol),摩尔收率87.47%(以化合物1:3,5-二氨基-2,4,6-三碘苯甲酸计算),hplc含
量99.3%。
[0046]
实施例7:泛影酸的精制
[0047]
重复实施例4的操作,产出泛影酸成品28.5g,质量收率93.75,总摩尔收率82%,hplc含量:99.98%,ep药典方法hplc图谱详见图2。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1