一种通过简并测序对目标样品定性分析或定量分析的方法与流程
1.本发明涉及一种通过简并测序对目标样品定性分析或定量分析的方法,属于基因测序技术领域。
背景技术:
2.目前,高通量测序技术广泛应用于对核酸分子的定性和/或定量分析,例如rna-seq、chip-seq、clip-seq、atac-seq等,通过对测序结果的分析得到目标核酸分子的有无或数量等信息。但是现有的方法必须先通过测序获得完整的碱基序列,再经过序列比对,以及生物信息学分析,最终得到目标核酸的信息。本发明提供一种方法,不需要先得到待测核酸的完整碱基序列信息,即可实现对目标样品的定性分析和/或定量分析。
技术实现要素:
3.本发明公开一种通过简并测序对目标样品定性分析或定量分析的方法,其特征在于,包括:
4.从目标样品中获得待测序核酸分子;对所述核酸分子构建的文库进行简并测序,得到初始序列;对所述初始序列和相应的参考序列用相同的编码方式分别进行编码,得到对应的编码后序列;将所述编码后序列进行比对得到比对结果;对所述比对结果利用生物信息学分析,得到目标样品的定性结果或定量结果;所述简并测序指的是3’端不封闭的测序反应;简并测序中,包括两种不同的测序试剂:第一测序试剂和第二测序试剂;两种测序试剂循环加入;其中所述第一测序试剂包含具有可检测标记的至少两种不同的核苷酸单体;所述第二测序试剂包含具有可检测标记的一种或多种核苷酸单体,且所述一种或多种核苷酸单体中的至少一种不同于所述第一测序试剂中存在的所述核苷酸单体,并且其中所述第二测序试剂是在提供了所述第一测序试剂随后提供的,将所述核苷酸单体掺入所述多核苷酸之后检测所述可检测标记生成的信号。
5.根据优选的实施方式,所述获得待测序核酸分子包括,rna提取获得待测rna分子、染色质免疫共沉淀技术获得待测dna分子、染色质转座酶可接近性实验获得待测dna、rna免疫共沉淀获得待测rna分子、染色体构象捕获技术获得的dna分子、微生物核酸成分的样品中提取的核酸分子、人外周血和/或胚胎培养液中提取获得的游离dna分子、dna编码化合物库筛选后提取的dna、核酸适配体体外筛选实验获得待测核酸分子等。
6.根据优选的实施方式,所述待测序核酸分子是dna分子或rna分子。
7.根据优选的实施方式,当待测序核酸分子为rna分子时,在构建文库前将rna逆转录为dna。
8.根据优选的实施方式,在测序之前对待测序核酸分子构建的文库进行扩增,所述扩增是pcr或恒温扩增的一种。
9.根据优选的实施方式,所述简并测序是2+2测序,指的是简并测序中,包括两种不同的测序试剂:第一测序试剂和第二测序试剂;两种测序试剂循环加入;其中所述第一测序
试剂包含具有可检测标记的两种不同的核苷酸单体;所述第二测序试剂包含具有可检测标记的两种核苷酸单体,且所述两种核苷酸单体不同于所述第一测序试剂中存在的所述核苷酸单体,并且其中所述第二测序试剂是在提供了所述第一测序试剂随后提供的,将所述核苷酸单体掺入所述多核苷酸之后检测所述可检测标记生成的信号。
10.根据优选的实施方式,所述简并测序是1+3测序,指的是简并测序中,包括两种不同的测序试剂:第一测序试剂和第二测序试剂;两种测序试剂循环加入;其中所述第一测序试剂包含具有可检测标记的三种不同的核苷酸单体;所述第二测序试剂包含具有可检测标记的一种核苷酸单体,且所述一种核苷酸单体不同于所述第一测序试剂中存在的所述核苷酸单体,并且其中所述第二测序试剂是在提供了所述第一测序试剂随后提供的,将所述核苷酸单体掺入所述多核苷酸之后检测所述可检测标记生成的信号。
11.根据优选的实施方式,所述两种测序试剂中的核苷酸单体5’多磷酸末端或中间磷酸修饰有荧光切换性质的荧光团;所述的荧光切换性质指的是测序后荧光信号相比测序反应前有明显改变。
12.根据优选的实施方式,所述的初始序列可以是使用m、k、r、y、w、s、b、d、h、v字母表示的简并碱基序列。
13.根据优选的实施方式,所述的初始序列可以是使用m、k、r、y、w、s、b、d、h、v字母表示的简并碱基序列和完整碱基序列的结合,所述的完整碱基序列指的是以a、g、t、c为编码的核酸序列信息,或者以a、g、u、c为编码的核酸序列信息;其中,碱基可以是甲基化的碱基。
14.根据优选的实施方式,所述编码指的是将测序获得的初始序列,改写成其可能的碱基序列信息中的一种。
15.根据优选的实施方式,所述参考序列是基因组或转录组或基因组的一个子集或转录组的一个子集。
16.根据优选的实施方式,所述比对可以通过使用bwa、bowtie2、tmap、eland、minimap2等软件完成。
17.根据优选的实施方式,所述定性分析包括,通过对所述比对结果进行生物信息学分析,确定目标样品中的微生物种类、确定目标样品中的单细胞的类型、确定染色质的层次结构、确定与目标蛋白质相互作用的dna或rna的种类等。
18.根据优选的实施方式,所述定量分析包括,转录组测序得到目标基因表达量、可变剪切、可变多聚腺苷酸化、基因表达速率等转录组信息;单细胞转录组测序得到目标单细胞的丰度、差异表达基因;chip-seq得到与目标蛋白质互作的dna的数量;clip-seq或rip-seq得到与目标蛋白质互作的rna的量;atac-seq得到染色质开放区域的dna的量;人外周血和/或胚胎培养液中的目标游离dna分子的数量。
19.本发明的有益效果
20.本发明的方法与现有技术相比,有以下优势:
21.1.可以只进行单轮简并测序,所得的序列信息足够用于比对,无需进行两轮或更多轮简并测序,节省了大量时间。
22.2.对于测序所得初始序列和对应的参考序列用相同的方式编码,独特的序列编码方式能够达到与常规测序的序列比对相近的效果。
23.3.应用广泛,可用于对多种生物样品进行定性或定量分析,包括组织或细胞的全
转录组、与特定蛋白质相互作用的dna分子、与特定rna结合蛋白相互作用的rna分子、染色质开放区域的dna分子、含有微生物核酸成分的样本等。
附图说明
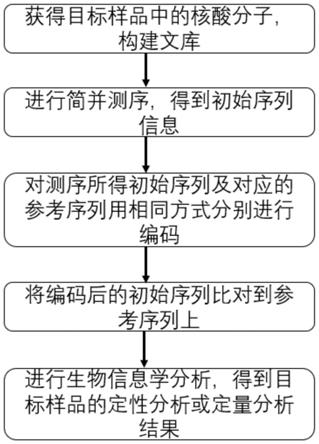
24.图1.本发明的方法的流程图。
25.图2.用三种测序方法进行全转录组测序所检测到的基因数目图。其中,bit seq表示单轮简并测序所得数据,illumina是用illumina miseq测序仪进行测序所得数据,ion proton是用thermofisher ion proton s5测序仪测序所得数据。
26.图3.三种测序方法所测tpm》1的基因的venn图。左图表示的是es细胞,右图表示的是mef。
27.图4.三种测序方法所得基因表达量的相关性图。
28.图5.三种测序方法所得的差异表达基因的表达量热图。
具体实施方式
29.以下结合附图和实施例对本发明作进一步说明。
30.除非另有定义,否则本文使用的所有科技术语具有与本领域普通技术人员通常理解的含义相同的含义。
31.如图1所示,本发明公开了一种通过简并测序对目标样品定性分析或定量分析的方法,其特征在于,包括:
32.从目标样品中获得待测序核酸分子;对所述核酸分子构建的文库进行简并测序,得到初始序列;对所述初始序列和相应的参考序列用相同的编码方式分别进行编码,得到对应的编码后序列;将所述编码后序列进行比对得到比对结果;对所述比对结果利用生物信息学分析,得到目标样品的定性结果或定量结果;所述简并测序指的是3’端不封闭的测序反应;简并测序中,包括两种不同的测序试剂:第一测序试剂和第二测序试剂;两种测序试剂循环加入;其中所述第一测序试剂包含具有可检测标记的至少两种不同的核苷酸单体;所述第二测序试剂包含具有可检测标记的一种或多种核苷酸单体,且所述一种或多种核苷酸单体中的至少一种不同于所述第一测序试剂中存在的所述核苷酸单体,并且其中所述第二测序试剂是在提供了所述第一测序试剂随后提供的,将所述核苷酸单体掺入所述多核苷酸之后检测所述可检测标记生成的信号。
33.根据优选的实施方式,获得待测序核酸分子包括,rna提取获得待测rna分子、染色质免疫共沉淀技术获得待测dna分子、染色质转座酶可接近性实验获得待测dna、rna免疫共沉淀获得待测rna分子、染色体构象捕获技术获得的dna分子、微生物核酸成分的样品中提取的核酸分子、人外周血和/或胚胎培养液中提取获得的游离dna分子、dna编码化合物库筛选后提取的dna、核酸适配体体外筛选实验获得待测核酸分子等。
34.本发明中的简并测序属于二代测序,在进行测序反应之前需要对待测核酸分子构建文库,优选的,需要对文库进行扩增,经过扩增得到足够的用于测序反应的核酸片段。扩增可以选择pcr或恒温扩增的一种,优选恒温扩增,包括,例如,扩增是环介导的等温扩增(lamp)、重组酶聚合酶扩增(rpa)、重组酶介导等温核酸扩增(raa)、切刻内切酶恒温扩增(near)、滚环扩增(rca)、依赖核酸序列的扩增(nasba)、解旋酶依赖性扩增(hda)、链置换扩
增(sda)、桥式扩增等。
35.根据优选的实施方式,所述两种测序试剂中的核苷酸单体5’多磷酸末端或中间磷酸修饰有荧光切换性质的荧光团;所述的荧光切换性质指的是测序后荧光信号相比测序反应前有明显改变;所述的明显改变可以是测序后荧光信号强度相比测序反应前有明显上升。
36.根据优选的实施方式,所述简并测序是2+2测序,指的是简并测序中,包括两种不同的测序试剂:第一测序试剂和第二测序试剂;两种测序试剂循环加入;其中所述第一测序试剂包含具有可检测标记的两种不同的核苷酸单体;所述第二测序试剂包含具有可检测标记的两种核苷酸单体,且所述两种核苷酸单体不同于所述第一测序试剂中存在的所述核苷酸单体,并且其中所述第二测序试剂是在提供了所述第一测序试剂随后提供的,将所述核苷酸单体掺入所述多核苷酸之后检测所述可检测标记生成的信号;其中所述第一测序试剂和所述第二测序试剂中的所述核苷酸单体选自由以下组合组成的组:
37.1)一种测序试剂中的腺嘌呤(a)核苷酸单体和胸腺嘧啶(t)/尿嘧啶(u)核苷酸单体以及另一种测序试剂中的胞嘧啶(c)核苷酸单体和鸟嘌呤(g)核苷酸单体;
38.2)一种测序试剂中的腺嘌呤(a)核苷酸单体和鸟嘌呤(g)核苷酸单体以及另一种测序试剂中的胞嘧啶(c)核苷酸单体和胸腺嘧啶(t)/尿嘧啶(u)核苷酸单体;和
39.3)一种测序试剂中的腺嘌呤(a)核苷酸单体和胞嘧啶(c)核苷酸单体以及另一种测序试剂中的鸟嘌呤(g)核苷酸单体和胸腺嘧啶(t)/尿嘧啶(u)核苷酸单体。
40.根据优选的实施方式,本发明的简并测序可以只使用上述一组核苷酸单体的组合进行测序,例如的:第一测序试剂包含a、c,第二测序试剂包含g、t,两种测序试剂循环加入,直至全部序列测序完成,此即一轮简并测序,或者叫单轮简并测序。根据iupac的核酸符号,用表1中的字母表示本发明中的简并碱基。例如,m代表a和/或c碱基,上述测序轮也可以简称为mk轮。
41.表1.表示简并碱基的字母
42.字母所代表的碱基ma/ckg/tra/gyc/twa/tsc/gbc/g/tda/g/tha/c/tva/c/g
43.根据优选的实施方式,所述简并测序是1+3测序,指的是简并测序中,包括两种不同的测序试剂:第一测序试剂和第二测序试剂;两种测序试剂循环加入;其中所述第一测序试剂包含具有可检测标记的三种不同的核苷酸单体;所述第二测序试剂包含具有可检测标记的一种核苷酸单体,且所述一种核苷酸单体不同于所述第一测序试剂中存在的所述核苷
酸单体,并且其中所述第二测序试剂是在提供了所述第一测序试剂随后提供的,将所述核苷酸单体掺入所述多核苷酸之后检测所述可检测标记生成的信号;其中所述第一测序试剂和所述第二测序试剂中的所述核苷酸单体选自由以下组合组成的组:
44.1)一种测序试剂中的胞嘧啶(c)核苷酸单体、鸟嘌呤(g)核苷酸单体和胸腺嘧啶(t)/尿嘧啶(u)核苷酸单体,以及另一种测序试剂中的腺嘌呤(a)核苷酸单体;
45.2)一种测序试剂中的腺嘌呤(a)核苷酸单体、鸟嘌呤(g)核苷酸单体和胸腺嘧啶(t)/尿嘧啶(u)核苷酸单体,以及另一种测序试剂中的胞嘧啶(c)核苷酸单体;和
46.3)一种测序试剂中的腺嘌呤(a)核苷酸单体、胞嘧啶(c)核苷酸单体和胸腺嘧啶(t)/尿嘧啶(u)核苷酸单体,以及另一种测序试剂中的鸟嘌呤(g)核苷酸单体;和
47.4)一种测序试剂中的腺嘌呤(a)核苷酸单体、胞嘧啶(c)核苷酸单体和鸟嘌呤(g)核苷酸单体,以及另一种测序试剂中的胸腺嘧啶(t)/尿嘧啶(u)核苷酸单体。
48.根据优选的实施方式,本发明的简并测序可以只使用上述一组核苷酸单体的组合进行测序,例如的:第一测序试剂包含c、g、t,第二测序试剂包含a,两种测序试剂循环加入,直至全部序列测序完成,此即一轮简并测序,或者叫单轮简并测序,上述测序轮也可以简称为ab轮。
49.需要注意的是,在现有技术中,以2+2简并测序为例,为了得到完整的碱基序列,需要进行至少2轮简并测序,才能得到以a,g,c,t(或u)表示的完整的碱基序列。而本发明中的方法只需要进行一轮简并测序,即单轮简并测序,即可得到用于比对的初始序列,节省了大量的测序时间和测序成本。
50.根据优选的实施方式,所述的初始序列信息可以是使用m、k、r、y、w、s、b、d、h、v字母表示的简并碱基序列。简并序列按照iupac符号命名规则(nucleic acid notation)。当进行单轮2+2简并测序时,若加入的底物分别是m和k,则得到的初始序列为由m、k表示的简并碱基序列;若单轮测序加入的底物分别是r和y,则得到的初始序列为由r、y表示的简并碱基序列;若单轮测序加入的底物分别是w和s,则得到的初始序列为由w、s表示的简并碱基序列。
51.根据优选的实施方式,所述的初始序列可以是使用m、k、r、y、w、s、b、d、h、v字母表示的简并碱基序列和完整碱基序列的结合。例如,对于1+3简并测序,只加入一种核苷酸底物的反应轮,此时发生延伸的碱基即为底物核苷酸,其序列是确定的,即完整碱基序列;而对于加入三种核苷酸底物的反应轮,此时发生的碱基延伸是无法准确确定其具体碱基序列的,属于简并碱基序列;因此,进行1+3简并测序得到的初始序列信息是简并碱基序列和完整碱基序列的结合。
52.根据优选的实施方式,对所得初始序列和相应的参考序列用相同的编码方式分别进行编码。所述编码指的是将测序获得的初始序列,改写成其可能的碱基序列信息中的一种。例如,对于mk单轮简并测序,m是a或c,则可以将m统一编码为a,也可以将m统一编码为c;对于k(g或t),则可以统一编码为g,或者统一编码为t,对于单轮mk测序所得的初始序列为mmkkkmkkmmm,则可以将其编码为aatttattaaa。相应的,对于参考序列,也将序列a、c都编码为a,序列g、t都编码为t,则可以得到编码后的参考序列。
53.本发明中,上述编码方法可以满足(也可以不满足)以下“编码与反向互补可交换”性质:对任意一条dna序列先编码、再作反向互补操作,或者先作反向互补操作、再编码,这
两种情况下得到的结果均相同。例如,对一条dna序列作单mk测序,规定编码方式为:将所测得的m全部改写为a,所测得的k全部改写为t。那么,对于序列acaggtg,该序列先编码后进行反向互补得到的序列为aaaattt;该序列先进行反向互补后编码得到的序列也是aaaattt,由此可见,这种编码方式是符合“编码与反向互补可交换”性质的。但如果规定编码方式为:将所测得的m全部改写为a,所测得的k全部改写为c。那么:对于序列acaggtg,该序列先编码后进行反向互补得到的序列为ggggttt;而对该序列先进行反向互补后编码得到的序列是aaaaccc,两种编码方式所得序列不相同,因此,不符合该“编码与反向互补可交换”性质。
54.如果选择的编码方式不符合“编码与反向互补可交换”性质,那么需要同时对参考基因组及其反向互补序列都进行编码,并同时将每条dna分子(编码后)的测序结果比对到其参考基因组及其反向互补序列的编码结果上,并从中选择一个较好的比对结果。
55.根据优选的实施方式,选择符合“编码与反向互补可交换”性质的编码方式进行编码,只需要对参考基因组进行编码,不需要对其反向互补序列也进行编码。
56.根据优选的实施方式,所述参考序列可以是基因组或转录组或基因组的一个子集或转录组的一个子集。参考序列是碱基序列分析时预先设定的序列,根据测序样品预先选择确定对应的参考序列,例如,测序的样品是人的病灶组织和正常组织,对其中的转录组进行测序,则选择的参考序列是人的转录组序列;测序的样品是经由染色质免疫共沉淀得到的dna分子,则选择对应物种的基因组作为参考序列。当只关注某个基因或某几个基因的表达情况时,例如进行肿瘤靶向测序时,通过探针或扩增的方式富集癌基因的dna,然后只对这些癌基因进行简并测序,其他和此癌症无关的dna都没有测序,此时可以把上述癌基因的序列摘取出来作为参考序列,即选择基因组的一个子集,而不需要选择整个基因组;在微生物鉴定中有类似的靶向测序的做法,例如只测16srdna,这样可以加快比对速度。
57.根据优选的实施方式,将测序序列比对到参考序列之后,便可以利用常规的生物信息学方法进行分析,此处所述的生物信息学方法包括但不限于,例如:筛选高质量的、可信的比对结果及对应的被比对的序列;统计比对到不同参考序列或参考序列的不同区域上的序列的条数或碱基数;根据gc含量、区域长度或其他序列特征,校正或归一化前述序列的条数或碱基数;利用前述校正或归一化后的序列条数或碱基数,针对具体的生物样品类型,进行统计推断或模型参数拟合。
58.可用于本发明此处的、常见的生物信息学工具包括但不限于,例如:对比对结果进行可视化的igv软件;对比对结果进行格式转换、筛选、合并、分割等操作的samtools软件;利用rna-seq测序结果对基因表达进行定量分析的salmon软件和star软件;利用rna-seq测序结果获得差异表达基因的r语言中的deseq2软件包;分析chip-seq测序结果的macs软件;r语言中的bioconductor软件包等。
59.以上软件均为常见的、本领域专业人员所熟知的生物信息学分析软件,此处不再赘述。
60.根据优选的实施方式,上述生物信息学分析可以得到关于目标样品的分析结果,包括定性分析和/或定量分析。
61.所述定性分析包括,通过对所述比对结果进行生物信息学分析,确定目标样品中的微生物种类、确定目标样品中的单细胞的类型、确定染色质的层次结构、确定与目标蛋白质相互作用的dna或rna的种类等。
62.根据优选的实施方式,所述定量分析包括,转录组测序得到目标基因表达量、可变剪切、可变多聚腺苷酸化、基因表达速率等转录组信息;单细胞转录组测序得到目标单细胞的丰度、差异表达基因;chip-seq得到与目标蛋白质互作的dna的数量;clip-seq或rip-seq得到与目标蛋白质互作的rna的量;
63.atac-seq得到染色质开放区域的dna的量;测序得到人外周血和/或胚胎培养液中的目标游离dna分子的数量。
64.根据优选的实施方式,目标样品核酸分子为转录组rna,则本发明的方法的具体实现步骤如下:
65.1.从目标样品中提取rna分子,逆转录为cdna,构建文库。
66.2.对文库进行扩增,进行单轮简并测序。
67.3.将测序获得的初始序列信息,编码成其可能的碱基序列信息中的一种。
68.4.将目标样品所属物种的参考转录组按相同方式编码为参考序列。
69.5.利用常用的转录组分析软件,将编码后的初始序列比对到编码后的参考序列上。
70.6.利用生物信息学方法计算基因表达量、可变剪切、可变多聚腺苷酸化等转录组信息。
71.根据优选的实施方式,步骤1中的文库构建方法可以是针对多个细胞的,也可以是针对单细胞的。针对单细胞的建库方法,包括但不限于:tang-protocol,smart-seq,smart-seq2,cel-seq,quartz-seq等。
72.根据优选的实施方式,步骤5所述转录组分析软件,包括但不限于:bowtie2,bwa-mem,star,salmon,gem,bbmap等。
73.根据优选的实施方式,步骤6中所述基因表达量,可以用rpkm、fpkm、tpm等形式表达。
74.根据优选的实施方式,当进行单细胞转录组测序(single cell rna sequencing)时,需要预先分离出单细胞,再对每个单细胞进行rna提取。单细胞转录组测序是在单细胞水平对转录组进行测序的一项技术,可以研究单个细胞内的基因表达情况,例如根据细胞是否表达某种或某几种特异性的标记因子,从而实现对细胞群体的划分等。在利用本发明的方法实现细胞群体划分的过程中,有时并不需要对基因进行精确定量,只需要根据此细胞是否有某种标记基因的表达,即可做出判断,这属于对目标样品的定性分析。当需要对细胞群体间基因表达差异进行检测时,需要对基因进行精确定量,此时实现的是对样品的定量分析功能。
75.根据优选的实施方式,待测核酸分子来源于含有微生物的样品,通过测序方法对微生物进行鉴定,利用本发明方法的具体实现步骤如下:
76.1.提取含有微生物核酸成分的样品中的核酸,构建文库。
77.2.对文库进行扩增,进行单轮简并测序。
78.3.将测序获得的初始序列信息,编码成其可能的碱基序列信息中的一种。
79.4.将候选微生物的参考基因组或转录组按相同方式编码为参考序列。
80.5.利用常用的序列比对软件和算法,将编码后的初始序列比对到编码后的参考序列上。
81.6.根据比对结果对所测微生物进行定性或定量分析。
82.根据优选的实施方式,步骤1中所述样品可以是土壤、大气、水体、粪便、动植物体表或体内中提取到的样品。
83.根据优选的实施方式,步骤1中所述的文库构建方式,既包括全基因组或全转录组的建库方式,也包括富集或靶向类的建库方式。特别的,步骤1中的文库构建方式,包括针对16s rrna的建库方式。在原核生物基因组中,编码16srrna的rdna基因具有良好的进化保守性、适宜分析的长度(约为1.5kb),以及与进化距离相匹配的良好变异性,所以成为细菌分子鉴定的标准标识序列。16s rdna具有10个保守区和9个可变区,其中保守区在细菌中差异不大,可变区具有属和种的特异性,对16s rdna可变区进行测序,可以用于研究环境微生物中细菌或古菌的群落多样性。
84.在某些实现中,样品中可能含有较多的来自于宿主的核酸。此时可以在步骤5之前增加一步:将编码后的初始序列比对到编码后的宿主参考基因组/转录组上,根据比对结果筛选一部分所测初始序列,再进行步骤5的操作。上述“根据比对结果筛选一部分所测初始序列”,可以是挑选未比对上的序列,或比对质量低的序列。
85.根据优选的实施方式,步骤5中所述常用的序列比对软件和算法,包括但不限于needleman-wunch算法、smith-waterman算法、blast、bowtie2、bwa-mem、tmap、eland、minimap2、novoalign等。
86.根据优选的实施方式,步骤6中所述“对所测微生物进行定性或定量分析”,可以指确定所测微生物的生物学分类、物种类型、菌株类型、突变体类型,也可以指所测微生物在样品中的丰度。
87.根据优选的实施方式,目标样品经由染色质免疫共沉淀得到待测dna分子,则本发明的方法的具体实现步骤如下:
88.1.由染色质免疫共沉淀获得待测序dna分子,进行文库构建;
89.2.对文库进行扩增,进行单轮简并测序;
90.3.将测序获得的初始序列信息,编码成其可能的碱基序列信息中的一种。
91.4.对参考基因组用相同编码方式进行编码,得到编码后的参考序列。
92.5.利用常用的序列比对软件和算法,将编码后的初始序列比对到编码后的参考序列上。
93.6.对比对上的dna分子进行定性或定量分析。
94.染色质免疫共沉淀测序技术(chip-seq)是将深度测序技术与chip实验相结合分析全基因组范围内dna结合蛋白结合位点、组蛋白修饰、核小体定位或dna甲基化的高通量方法,可以应用到任何基因组序列已知的物种。根据优选的实施方式,所述参考基因组为所测样品对应物种的基因组序列,可以从ensembl、ucsc、ncbi等渠道下载参考序列。
95.根据优选的实施方式,所述序列比对软件包括bwa、bowtie2、eland、soap等。
96.根据优选的实施方式,所述定性分析可以是根据序列比对结果得到与目标靶蛋白特异性结合的dna分子,更进一步的,所述定量分析是得到与靶蛋白结合的dna分子的数量。
97.类似地,本发明的方法还可以用于对与目标蛋白质互作的rna分子进行定性和定量分析,具体实现步骤如下:
98.1.通过免疫沉淀靶蛋白来捕获互作的rna分子,反转录为dna,进行文库构建。
99.2.对文库进行扩增,进行单轮简并测序。
100.3.将测序获得的初始序列信息,编码成其可能的碱基序列信息中的一种。
101.4.对参考转录组用相同编码方式进行编码,得到编码后的参考序列。
102.5.利用常用的序列比对软件,将编码后的初始序列比对到编码后的参考序列上。
103.6.利用生物信息学方法对比对到的rna分子进行定性或定量分析。
104.根据优选的实施方式,步骤1中所述免疫沉淀可以利用紫外交联免疫沉淀(clip)技术或者rna免疫共沉淀(rip)技术。紫外交联免疫沉淀测序(uv cross-linking and immunoprecipitation sequencing,clip-seq)用于研究rna结合蛋白在体内与众多rna靶标的结合模式,基于rna分子与rna结合蛋白在紫外照射下发生共价结合,提高结合强度,在全基因组范围内鉴定rna与rna结合蛋白的相互作用网络。rna immunoprecipitation sequencing(rip-seq)是rna免疫共沉淀结合高通量测序的一种技术,通过免疫沉淀靶蛋白来捕获与其互作的rna分子,将捕获的rna进行高通量测序,可在全基因组范围对蛋白结合位点进行筛选与鉴定;rip-seq与clip-seq相比,不需要进行紫外交联,操作简单,成功率更高。
105.根据优选的实施方式,所述rna分子包括mrna,mirna,lncrna,circrna等。
106.根据优选的实施方式,本发明中的目标样品可以是人外周血和/或胚胎培养液,对上述样品内的游离dna分子进行高通量测序,可以为癌症的无创诊断,特别是早期诊断提供新的辅助手段,同时也是一种非侵入性的产前筛查方式。具体实现步骤如下:
107.1.分离血液或胚胎培养液中的游离dna分子,进行文库构建。
108.2.对文库进行扩增,进行单轮简并测序。
109.3.将测序获得的初始序列信息,编码成其可能的碱基序列信息中的一种。
110.4.对参考基因组用相同编码方式进行编码,得到编码后的参考序列。
111.5.利用常用的序列比对软件,将编码后的初始序列比对到编码后的参考序列上。
112.6.对比对上的dna分子进行定性或定量分析。
113.根据优选的实施方式,根据步骤6中的分析结果,对游离dna来源个体的生理状态作出判断,例如,若样品是孕妇的外周血或胚胎的培养液,则可以根据定量分析结果准确判断胎儿是否患有唐氏综合征(t21)、爱德华综合征(t18)、帕套综合征(t13)三大染色体疾病。
114.概括来讲,本发明的方法,适用范围广泛,对于需要通过核酸测序对目标样品进行分析的实验,无论是定性分析还是定量分析,本发明的方法都能够满足需求。
115.以下描述根据上述描述进行的实施例。
116.实施例1
117.本实施例描述了利用本发明的方法以及两种现有方法对两种细胞系分别进行全转录组测序、序列比对以及定量分析的过程。
118.体外培养小鼠胚胎成纤维细胞系(mef)和胚胎干细胞系(es),提取rna后反转录为cdna,构建文库;对文库进行扩增;分别使用illumina miseq测序仪、thermofisher ion proton s5测序仪、单轮简并测序方法进行测序,前二者为商业化测序仪,遵循生产商提供的标准操作和分析流程获得每个基因的表达量;其中简并测序为mk轮测序,得到初始序列,并将m编码为a、k编码为t。将小鼠的参考转录组中的所有a和c均编码为a,所有g和u均编码
为t;再用软件salmon将简并测序后编码得到的序列比对到编码后的参考转录组上。软件salmon可自动以tpm的形式计算出基因表达量。
119.在es和mef两种细胞中,三种测序方法所测基因表达量均表现出极高的线性相关性(图4)。分别以tpm》1和tpm》0.1为检测到某基因表达的阈值,三种测序方法在es和mef两种细胞中所检测到表达的基因数目也相互接近(图2),bit seq表示单轮简并测序。用venn图分析了三种测序方法所测tpm》1的基因种类,发现相互之间有很大的重叠(图3),其中es细胞检测到9812个基因、mef细胞有10545个基因被三种测序方法所同时检测到。进一步比较了es细胞和mef细胞的基因表达谱,并使用r语言的deseq2包作了差异表达分析。一共得到1438个表达量相差4倍以上、p value小于0.01的差异表达基因,其表达量的热度图显示三种测序方法所得差异表达基因之间也有极高的一致性(图5)。这些结果表明三种测序方法在转录组测定上取得了高度一致的结果。
120.以上对比实施例证明本发明的方法能够实现与现有技术相近的测序、比对及分析水平,而且只需进行一轮简并测序,不需要得到完整的碱基序列,因此,具有更明显的优势。
121.对所公开的实施例的上述说明,使得本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其他实施例中实现。因此,本发明将不会被限制于本文所示的实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1