一种莫那比拉韦的合成方法与流程
1.本发明属于药物合成领域,具体涉及一种莫那比拉韦的合成方法。
技术背景
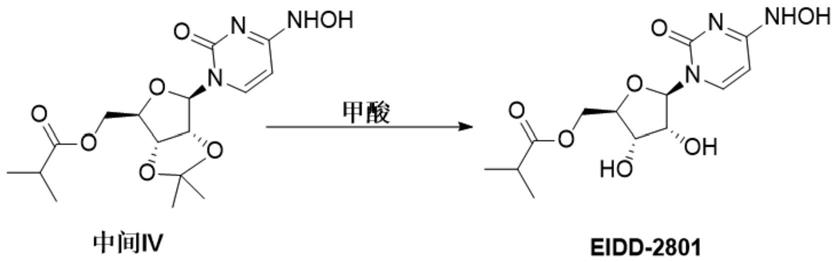
2.莫那比拉韦(eidd-2801,mk-4482)是一种由美国药业巨头默沙东研发的一种小分子胞苷类抗病毒药物,具有广谱的抗病毒活性对抗sars-cov-2、mers-cov、sars-cov和covid-19的病原体。因为口服药物使用方便,既可以作为预防用药,也适用于门诊病人和住院病人,其结构式如下:
[0003][0004]
目前莫那比拉韦的主要合成路线为专利wo2019113462,wo2019173602报道的化合物合成路线如下:
[0005][0006]
此步路线中,使用约18.8倍质量比的甲酸,后处理需要把大大过量的甲酸浓缩出去;另外专利cn112552288a中提到了可以是甲酸、乙酸、三氟乙酸、hcl中的一种,但是其案例中仍是使用4倍质量比的甲酸,后处理仍是需要浓缩过量的甲酸;
[0007]
在实验室阶段甲酸投料量相对少,浓缩甲酸时间较短,但在生产中投料量大,浓缩甲酸时间长,且在浓缩过程中,主产物减少,杂质a、杂质b逐渐增大,另外反应中也有杂质c 的生成,副产物较多给产品的纯化带来一定的困难,需要多次纯化才可以得到高纯度产品,总的收率也降低。此外,随着生产批量的放大,浓缩甲酸不仅时间长,而且对设备的要求也越来越高。
[0008]
专利in202141011933中公开了采用盐酸或三氟乙酸脱缩醛,重复此工艺,发现原料剩余较多,且由于采用过量的强酸,导致杂质c较大。
[0009]
所述的杂质a、杂质b、杂质c的结构如下:
[0010][0011]
因此,在该脱缩醛制备步骤中,使用远远过量的甲酸、乙酸等弱酸,不仅产生杂质a、杂质b,而且在大生产中长时间的浓缩甲酸时,杂质a和b会有明显的放大效应;使用盐酸、三氟乙酸等强酸,虽然未有杂质a、杂质b生产,但是由于酸性强,产品水解较快,导致杂质c较大,收率低。
技术实现要素:
[0012]
本发明为解决上述问题,提供了一种莫那比拉韦的合成方法,采用混酸,避开了浓缩甲酸、乙酸等,并通过控制强酸的用量进而控制杂质c的水解大小,该方法反应时间短,产生的杂质大大减少,主产物产率高,操作简便,更易于工业化生产及得到高纯度的产品。
[0013]
本发明提供的莫那比拉韦的合成方法具体包括如下步骤:
[0014][0015]
向反应瓶中加入莫那比拉韦中间体ⅳ和水、盐酸、甲酸,升温至50~75℃反应,优选 70~75℃,反应完全后,降温至室温,加入碱及有机溶剂i萃取,水相用有机溶剂i萃取,有机相经水洗涤,将有机相浓缩得粗品,粗品加入有机溶剂ii升温结晶,降温至室温,过滤,湿品真空干燥后得到成品莫那比拉韦。
[0016]
在一些实施方案中,所述碱选自碳酸钠,碳酸钾,碳酸氢钾,碳酸氢钠中的一种;
[0017]
在一些实施方案中,所述有机溶剂i选自二氯甲烷,乙酸乙酯,四氢呋喃,醋酸异丙酯和2-甲基四氢呋喃中的一种;
[0018]
在一些实施方案中,所述有机溶剂ii选自无水乙醇,乙醇和异丙醇中的一种。
[0019]
在一些实施方案中,中间体ⅳ∶水∶盐酸:甲酸的质量比为1∶(1~10)∶(0.10~0.26)∶(0.12~0.25),优选1∶3:0.10∶0.25。
[0020]
在一些实施方案中,水洗涤的次数为3~10次,优选4次;
[0021]
本发明中莫那比拉韦中间体ⅳ以尿苷为起始原料,先经丙酮叉保护双羟基,然后与异丁酸酐完成酯化反应,经缩合,再与羟胺反应得到的。
[0022]
有益效果:与现有技术相比,本发明提供的莫那比拉韦的合成方法,物料使用量少,反应时间短,产率高,产品纯度高,对设备要求低,操作简单,更易于工业化生产。
附图说明
[0023]
图1为实施例1中控取样检测hplc图谱;
[0024]
图2为实施例1浓缩后取样检测hplc图谱;
[0025]
图3为实施例1一次精制后取样检测hplc图谱;
[0026]
图4为实施例1二次精制后取样检测hplc图谱;
[0027]
图5为实施例2中控取样检测hplc图谱;
[0028]
图6为实施例3中控取样检测hplc图谱;
[0029]
图7为实施例4中控取样检测hplc图谱;
[0030]
图8为实施例5反应1取样检测hplc图谱;
[0031]
图9为实施例5反应2取样检测hplc图谱;
[0032]
图10为实施例5反应3取样检测hplc图谱;
[0033]
图11为实施例5反应4取样检测hplc图谱;
[0034]
图12为实施例5反应5取样检测hplc图谱;
[0035]
图13为实施例5反应6取样检测hplc图谱;
[0036]
图14为实施例5反应7取样检测hplc图谱;
[0037]
图15为实例6中控取样检测hplc图谱;
[0038]
图16为实例6成品取样检测hplc图谱;
具体实施方式
[0039]
以下为本发明的具体实施方式,所述的实施例是为了进一步描述本发明,而不是限制本发明。
[0040]
实施例1
[0041]
向反应釜中加入50kg莫那比拉韦中间体ⅳ,50kg纯化水,开启搅拌,加入250kg无水甲酸,升温至50~60℃反应至完全,取样检测,浓缩甲酸,取样检测;向浓缩物中抽入250kg 异丙醇,升温至溶清,降温,离心,干燥得到30.70kg莫那比拉韦第一次精制品,纯度89.93%。向反应釜中加入30.70kg莫那比拉韦第一次精制品,抽入250kg异丙醇,升温至体系溶清,降温析出,离心,湿品干燥得到27.42kg莫那比拉韦第二次精制品,纯度98.95%,收率61.50%。
[0042]
实验中我们发现,莫那比拉韦杂质a、杂质b在浓缩此过程中,明显变大,一次精制后纯度较低,需要二次精制;随着生产批量的放大,浓缩甲酸对设备和浓缩时间有着越来越高的要求,对产品质量也存在风险。
[0043]
实施例2
[0044]
向反应瓶中加入10g莫那比拉韦中间体ⅳ、25g 20%氯化氢乙醇溶液,开启搅拌,升温至 50~55℃,50~55℃反应3小时,取样检测发现无杂质a、杂质b的生成,但是中间体ⅳ原料剩余较多,此时杂质c较大,无明显优势。
[0045]
实施例3
[0046]
向反应瓶中加入10g莫那比拉韦中间体ⅳ、30g水,开启搅拌,加入10g三氟乙酸,升温至50~55℃,50~55℃反应3小时,取样检测发现无杂质a、杂质b的生成,但是中间体ⅳ原料剩余较多,且杂质c较大,无明显优势。
[0047]
实施例4
[0048]
向反应瓶中加入10g莫那比拉韦中间体ⅳ、30g水,开启搅拌,加入3g硫酸氢钾,升温至50~55℃,50~55℃反应3小时,中控取样检测发现无杂质a、杂质b的生成,但是中间体ⅳ原料剩余略多,且杂质c较大。
[0049]
表一:实施例1-4的检测数据(各组份的峰面积[mau*s]百分比),各取样点检测结果见图1~图7。
[0050]
取样点杂质c莫那比拉韦杂质a+杂质b中间体ⅳ实例1中控0.4486.659.692.80实例1浓缩后0.1269.3023.613.71实例1一次精制0.2190.165.623.07实例1二次精制0.0599.280.260.41实例2中控13.5962.140.0023.80实例3中控7.8255.770.0034.95实例4中控13.6178.910.005.25
[0051]
实施例5
[0052]
向反应瓶中加入莫那比拉韦中间体ⅳ、水、盐酸、甲酸,开启搅拌,升温至一定温度,在该温度下反应1-2h,取样检测,具体投料量及反应温度按下表设置:
[0053]
表二:投料量、反应温度对反应的影响,各反应取样检测结果见图8~图14。
[0054][0055]
从上表发现,中间体ⅳ∶盐酸∶甲酸∶水为1∶0.10∶0.25∶3,反应温度为70-75℃时,反应液中,莫那比拉韦组份最高,因此生产选择此反应条件进行放大验证;
[0056]
实施例6
[0057]
向反应釜中加入50kg莫那比拉韦中间体ⅳ和150kg水,开启搅拌,加入5kg盐酸、12.5kg 甲酸,升温至70~75℃,70~75℃反应1h,取样检测,降温,加入40.67kg碳酸氢钾、200kg2-甲基四氢呋喃萃取,水相用200kg 2-甲基四氢呋喃萃取,有机相用水洗涤4次,每次50kg 水,浓缩,加入250kg异丙醇,升温至溶清,降温,离心,湿品干燥得到37.11kg莫那比拉韦成品,纯度99.93%,收率83.26%。
[0058]
实验中我们发现,中控无杂质a、杂质b的生成,中间体ⅳ原料剩余较少,此时杂质c 为8.25%,但是杂质c有较好的水溶性,通过水洗涤可以去除,一次精制后得到的产品符合质量标准。取样检测结果见图15~图16。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1