一种索非布韦杂质的制备方法与流程
1.本发明涉及一种化合物的制备方法,尤其涉及一种索非布韦杂质的制备方法。
背景技术:
2.索非布韦(又译为索氟布韦,英文名sofosbuvir,商品名sovaldi)是吉利德公司开发用于治疗慢性丙肝的新药,于2013年12月6日经美国食品药品监督管理局(fda)批准在美国上市,2014年1月16日经欧洲药品管理局(emea)批准在欧盟各国上市。该药物是首个无需联合干扰素就能安全有效治疗某些类型丙肝的药物。临床试验证实针对1和4型丙肝,该药物联合聚乙二醇干扰素和利巴韦林的总体持续病毒学应答率(svr)高达90%;针对2型丙肝,该药物联合利巴韦林的svr为89%-95%;针对3型丙肝,该药物联合利巴韦林的svr为61%-63%。
3.随着时代的进步、科技水平的提高,人们对药品上市前必须对药品进行质量、安全性和效能科学评价的重要性等有了更加充分的认识,其中与药品质量密切相关的是药物所含杂质的控制。杂质往往与药品安全性有关,且在少数情况下与效能也有关。因此,控制杂质水平在药物开发研究过程中越来越受到医药工作者的重视。在索非布韦的合成和储存过程中会产生这个杂质,尚未见该杂质公开合成方法。
技术实现要素:
4.发明目的:本发明旨在提供一种工艺设计合理的索非布韦杂质的制备方法。
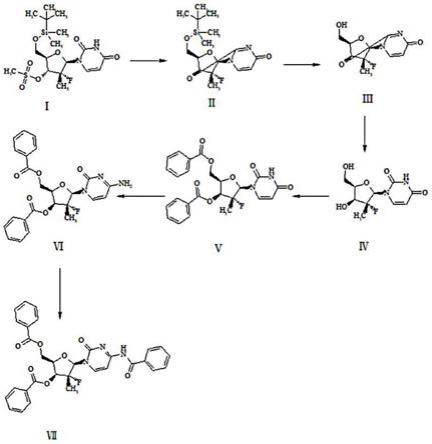
5.技术方案:本发明所述的索非布韦杂质的制备方法,包括以下步骤:(1) 取中间体ⅰ作为原料,用甲苯作溶剂,加入碱,反应得到中间产物ⅱ;(2) 取步骤(1)制备得到的中间产物ⅱ,用甲醇作溶剂,加入酸,反应生成中间产物ⅲ;(3) 取步骤(2)得到的中间产物ⅲ溶解在甲醇溶剂中,加入碱,反应得到中间产物ⅳ;(4) 取步骤(3)得到的中间产物ⅳ溶解在四氢呋喃中,加入碱,再加入苯甲酰氯,反应得到产物
ⅴ
;(5) 取步骤(4)得到的中间产物
ⅴ
溶解在四氢呋喃中,加入吡啶,再加入甲基磺酰氯,然后加入碱,反应得到产物ⅵ;(6) 取步骤(5)得到的中间产物ⅵ溶解在乙腈中,加入碱,再加入苯甲酰氯,反应得到最终产物ⅶ;该制备过程的反应式如下:
。
6.优选地,步骤(1)所述中间体ⅰ与碱的摩尔用量比为1:1~1:10;所述碱为氢化钠、氢氧化锂、氢氧化钠、氢氧化钾或者三乙胺;所述反应时间为6-24小时。
7.优选地,步骤(1)所述中间体ⅰ与碱的摩尔用量比为1:3~1:5;所述碱为三乙胺;所述反应时间为10-16小时。
8.优选地,步骤(2)所述的中间产物ⅱ与酸的摩尔用量比为1:1~1:6;所述酸为盐酸、硫酸、甲酸或者乙酸;所述反应时间为1-16小时。
9.优选地,步骤(2)所述的中间产物ⅱ与酸的摩尔用量比为1:2~1:4;所述酸为乙酸;所述反应时间为1-10小时。
10.优选地,步骤(3)所述的中间产物ⅲ与碱的摩尔用量比为1:1~1:8;所述碱为氢氧化钾、氢氧化钠、氢氧化锂、三乙胺、叔丁醇钠或者叔丁醇钾;所述反应时间为2~24小时。
11.优选地,步骤(3)所述的中间产物ⅲ与碱的摩尔用量比为1:3~1:5;所述碱为叔丁醇钾;所述反应时间为8~16小时。
12.优选地,步骤(4)所述的中间产物ⅳ与碱的摩尔用量比为1:2~1:8;步骤(4)所述的中间产物ⅳ与苯甲酰氯的摩尔用量比为1:2~1:10;所述碱为三乙胺、吡啶或者碳酸钾;所述反应时间为2~20小时。
13.优选地,步骤(4)所述的中间产物ⅳ与碱的摩尔用量比为1:2~1:5;步骤(4)所述的
中间产物ⅳ与苯甲酰氯的摩尔用量比为1:2~1:4;所述碱为三乙胺;所述反应时间为8~16小时。
14.优选地,步骤(5)所述的中间产物
ⅴ
与碱的摩尔用量比为1:2~1:12;步骤(5)所述的中间产物
ⅴ
与甲基磺酰氯的摩尔用量比为1:1~1:6;所述碱为氨水、含氨甲醇或者含氨乙醇;所述反应时间为6~20小时。
15.优选地,步骤(5)所述的中间产物
ⅴ
与碱的摩尔用量比为1:4~1:6;步骤(5)所述的中间产物
ⅴ
与甲基磺酰氯的摩尔用量比为1:2~1:4;所述碱为氨水;所述反应时间为8~16小时。
16.优选地,步骤(6)所述的中间产物ⅵ与碱的摩尔用量比为1:2~1:10;所述碱为碳酸钾、叔丁醇钾或者三乙胺;所述反应时间为4~24小时。
17.优选地,步骤(6)所述的中间产物ⅵ与碱的摩尔用量比为1:3~1:6;所述碱为碳酸钾;所述反应时间为8~14小时。
18.有益效果:与现有技术相比,本发明具有如下显著优点:该方法工艺设计合理,操作方法简单、原料易得、纯度高、反应过程可控和环境保护效果好,根据该方法制备得到的索非布韦杂质为索非布韦的研究提供测试样品,在临床药代动力学研究中具有重要研究价值,有利于对索非布韦临床、药理、药代动力学,毒理进行全面的分析、研究。该制备方法为全面分析索非布韦的临床、药理、药代动力学,毒理提供分析研究的基准物质。
附图说明
19.图1为本发明提供的索非布韦杂质的制备工艺流程图;图2为本发明提供的索非布韦杂质的最终产物核磁谱图。
具体实施方式
20.下面结合附图对本发明的技术方案作进一步说明。
21.实施例1索非布韦杂质的制备工艺流程图如图1所示。
22.化合物ⅱ的制备:将20g中间体ⅰ溶于200 ml甲苯,加入13.4g三乙胺,25度反应12小时,反应结束,反应液旋干,粗产物用二氯甲烷和甲醇体系过柱提纯得到14.5g黄色固体ⅱ,收率为92%。
23.化合物ⅲ的制备:取14.5g化合物ⅱ溶解到145ml甲醇中,室温下加入4.9g乙酸,80度反应5小时,tlc监控反应结束,将反应液旋干,加入200ml水,冰浴下加入饱和碳酸氢钠调ph=8,用二氯甲烷萃取三次,每次50ml,有机层合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用二氯甲烷和甲醇体系过柱提纯得到9g白色固体ⅲ,收率为91%。
24.化合物ⅳ的制备:取9g化合物ⅲ溶于90ml甲醇溶剂中,加入12.5g叔丁醇钾,反应液50度反应12小时,tlc监控反应结束,加入1m hcl调ph=7,反应液旋干,粗产物用二氯甲烷和甲醇体系过柱提纯得到8.5g白色固体ⅳ,收率为88%。
25.化合物
ⅴ
的制备:取8.5g化合物ⅳ溶于50ml四氢呋喃,加入6.6g 三乙胺,再加入18.3g苯甲酰氯,60度反应12小时,原料反应完,冰浴下加入100ml水,然后用乙酸乙酯萃取三次,每次100ml,有机相合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用乙醚重结
晶得到10.0g白色固体
ⅴ
,收率为82%。
26.化合物ⅵ的制备:取5g化合物
ⅴ
溶于四氢呋喃,加入2g吡啶,再加入2.4g甲基磺酰氯,50度反应16小时,降温到室温,再加入6g氨水,50度反应12小时,原料反应完,冰浴下加入1m hcl调ph=7,加入100ml水,然后用乙酸乙酯萃取三次,每次100ml,有机相合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用二氯甲烷和甲醇体系过柱提纯得到4.3g白色固体ⅵ,收率为86%。
27.化合物ⅶ的制备:取4g化合物ⅵ溶于乙腈,加入3.5g碳酸钾,再加入1.5g苯甲酰氯,60度反应10小时,原料反应完,反应液旋干,加入80ml水,然后用乙酸乙酯萃取三次,每次约80ml,有机相合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用乙酸乙酯和正己烷体系过柱提纯得到4.0g白色固体ⅶ,收率为81.7%,hplc:99.37%。核磁谱图见图2 (dmso-d6)。
28.实施例2化合物ⅱ的制备:将15g中间体ⅰ溶于150 ml甲苯,加入33.5g三乙胺,25度反应12小时,反应结束,反应液旋干,粗产物用二氯甲烷和甲醇体系过柱提纯得到9g黄色固体ⅱ,收率为76%。
29.化合物ⅲ的制备:取9g化合物ⅱ溶解到90ml甲醇中,室温下加入50.5ml 1m hcl,80度反应5小时,tlc监控反应结束,将反应液旋干,冰浴下加入饱和碳酸氢钠调ph=8,用二氯甲烷萃取三次,每次100ml,有机层合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用二氯甲烷和甲醇体系过柱提纯得到5g白色固体ⅲ,收率为81.7%。
30.化合物ⅳ的制备:取5g化合物ⅲ溶于50ml甲醇溶剂中,加入3.5g氢氧化钾,反应液50度反应12小时,tlc监控反应结束,加入1m hcl调ph=7。反应液旋干,粗产物用二氯甲烷和甲醇体系过柱提纯得到4g白色固体ⅳ,收率为74.4%。
31.化合物
ⅴ
的制备:取4g化合物ⅳ溶于40ml四氢呋喃,加入2.4g吡啶,再加入8.6g苯甲酰氯,60度反应12小时,原料反应完,冰浴下加入100ml水,然后用乙酸乙酯萃取三次,每次100ml,有机相合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用乙醚重结晶得到4.1g白色固体
ⅴ
,收率为71%。
32.化合物ⅵ的制备:取4g化合物
ⅴ
溶于四氢呋喃,加入1.5g吡啶,再加入2g甲基磺酰氯,50度反应16小时,降温到室温,再加入10ml含氨甲醇,50度反应12小时,原料反应完,冰浴下加入1m hcl调ph=7,加入100ml水,然后用乙酸乙酯萃取三次,每次100ml,有机相合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用二氯甲烷和甲醇体系过柱提纯得到3.1g白色固体ⅵ,收率为77.6%。
33.化合物ⅶ的制备:取3g化合物ⅵ溶于乙腈,加入1.9g三乙胺,再加入1.2g苯甲酰氯,60度反应10小时,原料反应完,反应液旋干,加入70ml水,然后用乙酸乙酯萃取三次,每次约70ml,有机相合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用乙酸乙酯和正己烷体系过柱提纯得到2.8g白色固体ⅶ,收率为76.3%。
34.实施例3化合物ⅱ的制备:将20g中间体ⅰ溶于300 ml 甲苯,加入4.5g 三乙胺,25度反应12小时,反应结束,反应液旋干,粗产物用二氯甲烷和甲醇体系过柱提纯得11g黄色固体ⅱ,收率为69.8%。
35.化合物ⅲ的制备:取11g化合物ⅱ溶解到110ml甲醇中,室温下加入11.1g 乙酸,80度反应3小时,tlc监控反应结束,将反应液旋干,冰浴下加入饱和碳酸氢钠调ph=8,用二氯甲烷萃取三次,每次80ml,有机层合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用二氯甲烷和甲醇体系过柱提纯得到5.5g白色固体ⅲ,收率为73.6%。
36.化合物ⅳ的制备:取5g化合物ⅲ溶于50ml甲醇溶剂中,加入6g叔丁醇钠,反应液50度反应10小时,tlc监控反应结束,加入1m hcl调ph=7,反应液旋干,粗产物用二氯甲烷和甲醇体系过柱提纯得到4.3g白色固体ⅳ,收率为80%。
37.化合物
ⅴ
的制备:取4g化合物ⅳ溶于40ml四氢呋喃,加入12.4g三乙胺,再加入8.6g苯甲酰氯,60度反应10小时,原料反应完,冰浴下加入80ml水,然后用乙酸乙酯萃取三次,每次80ml,有机相合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用乙醚重结晶得到4.5g白色固体
ⅴ
,收率为78%。
38.化合物ⅵ的制备:取4.5g化合物
ⅴ
溶于四氢呋喃,加入1.5g吡啶,再加入6.6g甲基磺酰氯,50度反应12小时,降温到室温,再加入12ml含氨甲醇,50度反应12小时,原料反应完,冰浴下加入1m hcl调ph=7,加入120ml水,然后用乙酸乙酯萃取三次,每次100ml,有机相合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用二氯甲烷和甲醇体系过柱提纯得到3.4g白色固体ⅵ,收率为75%。
39.化合物ⅶ的制备:取2g化合物ⅵ溶于乙腈,加入4.3g三乙胺,再加入0.8g苯甲酰氯,60度反应6小时,原料反应完,反应液旋干,加入50ml水,然后用乙酸乙酯萃取三次,每次约50ml,有机相合并用无水硫酸钠干燥,过滤,滤液旋干得到粗产物,用乙酸乙酯和正己烷体系过柱提纯得到1.8g白色固体ⅶ,收率为73.6%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1