地塞米松环氧水解物的精制方法与流程
1.本发明涉及甾体的提纯精制领域,更具体地,本发明涉及一种用于精制地塞米松环氧水解物的方法。
背景技术:
2.8dm(9β,11β-epoxy-17,21-dihydroxy-16α-methylpregna-1,4-diene-3,20-dione),也称为地塞米松环氧水解物,是甾体药物化学的重要中间体之一,8dm的cas号为:24916-90-3,分子式为:c
22h28
o5,分子量为:372.46,其结构式如下所示:
[0003][0004]
8dm是合成地塞米松和地塞米松系统产品的重要中间体。地塞米松系列产品作为非常重要的皮质激素,在临床上广泛应用于治疗皮肤病、风湿性疾病、严重过敏、哮喘等。
[0005]
目前,8dm生产工艺的最后一步多采用发酵工艺,通过发酵工艺得到的8dm纯度不高,需要进行进一步精制。现有技术均采用两溶剂体系的精制方法,经过精制后的8dm中仍存在较大量的未知的单个杂质,且杂质的结构复杂,难以确证进行结构确证,不利于杂质研究。
[0006]
专利公布文本cn110157764b中提到了8dm的提取精制条件,将8dm粗品加入到精制瓶中,采用20v-30v二氯甲烷/丙酮加热,回流30分钟后,减压浓缩至1v-3v丙酮,降温到20℃以下,进行抽滤获得湿精品,湿精品干燥,获得8dm精品。上述精制方法选择了二氯甲烷/丙酮的混合溶剂作为溶剂,其中二氯甲烷和丙酮混合比例为4∶1至4∶3;得到的8dm精品hplc纯度≥99.0%,单个杂质均小于0.5%,该方法纯化得到的8dm的单杂较大,作为原料药的起始物料进行注册申报的风险较大。
[0007]
专利申请公开文本cn109369768a实施例中8dm粗品在二氯甲烷和甲醇体系蒸馏除去二氯甲烷,降温至-10℃,保温3-5小时后过滤,洗涤,于35℃真空干燥,制备得到的8dm纯度最高可达98.7%,未提供单杂情况。
[0008]
专利公布文本cn108864240b提供了一种地塞米松环氧水解物的提纯方法,该方法包括如下步骤:将含8dm的母液浓缩至干后,溶于酯类溶剂中,得混合液a;将所述混合液a和无机钙盐的醇溶液混合发生络合反应,过滤,得8dm络合物;将所述8dm络合物依次经水洗和干燥处理,得到8dm粗品。所述8dm粗品经精制后得到8dm精品。通过该方法可实现母液中8dm的分离提纯,回收所得的8dm纯度达到93%以上,最高可达98.5%以上,未提供单杂情况。
[0009]
专利申请公开文本cn108395465a公开了一种地塞米松环氧水解物的合成方法,以氯仿、甲醇作为精制溶剂,得到的8dm纯度≥99%,单杂≤0.3%。
[0010]
通过现有技术中的精制方法获得的8dm中均存在大于0.1%的杂质,而在原料药注册申报过程中大于0.1%的杂质需要进行杂质研究,不利于以8dm作为起始物料来制备的原
料药的注册申报。
[0011]
因此,本领域亟需一种地塞米松环氧水解物(8dm)的精制方法,该方法可以去除目前主流的8dm发酵工艺和化学合成工艺质中较大量的未知单杂和已知单杂,提供高纯度的8dm,以利于8dm作为起始物料的原料药进行注册申报。
技术实现要素:
[0012]
如上所述,现有的地塞米松环氧水解物的精制方法,制备获得的地塞米松环氧水解物纯度不够高,含有大于0.1%的单个杂质。因此,本领域亟需一种地塞米松环氧水解物的精制方法,可以去除8dm中大于0.1%的单个杂质,提供高纯度的8dm。
[0013]
因此,在第一方面,本发明提供了一种地塞米松环氧水解物的精制方法,其特征在于,所述精制方法包括以下步骤:
[0014]
1)将地塞米松环氧水解物粗品溶于第一溶剂、第二溶剂和第三溶剂的混合物中,并升温至体系溶清得到第一体系,其中,所述第一溶剂为四氢呋喃或取代的四氢呋喃,所述第二溶剂为低级醇,所述第三溶剂为水,并且所述第一溶剂、所述第二溶剂、所述第三溶剂与所述地塞米松环氧水解物粗品的体积质量比以ml/g计为(3-5):(3-8):(0.1-1):1;
[0015]
2)将所述第一体系蒸馏得到第一固液混合体系,作为第二体系;
[0016]
3)将所述第二体系于第一温度搅拌析晶得到第二固液混合体系,作为第三体系,其中,所述第一温度为20℃至80℃,所述搅拌析晶的时间为1小时至3小时;
[0017]
4)将所述第三体系于第二温度搅拌析晶得到第三固液混合体系,作为第四体系,其中,所述第二温度为-5℃至5℃,所述搅拌析晶的时间为3小时至5小时;
[0018]
5)将所述第四体系进行固液分离以得到固体产物。
[0019]
在第二方面,本发明提供了一种精制的地塞米松环氧水解物,其是通过本发明第一方面所述的精制方法制备得到的,其中所述地塞米松环氧水解物的纯度≥99.5%,并且其包含的单个杂质均小于0.1%。
[0020]
本发明的有益效果为:提供了一种地塞米松环氧水解物的精制方法,可有效去除8dm粗品中的杂质,获得纯度达到99.5%以上,单个杂质小于0.1%,总杂质小于0.5%的8dm,同时保证8dm精品的收率达到90%以上。所述方法具有收率高、产品质量好、处理过程中体系稳定、操作简单、易于实现工业化生产等优势。
附图说明
[0021]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的实施方案。
[0022]
图1为实施例4中8dm精品的hplc检测图谱。
[0023]
图2为实施例5中8dm精品的hplc检测图谱。
[0024]
图3为实施例6中8dm精品的hplc检测图谱。
具体实施方式
[0025]
下面将结合本发明的实施方案和附图,对本发明进行清楚、完整的描述。显然,所描述的实施方案仅仅是本发明的一部分实施方案,而不是全部的实施方案。基于本发明中的实施方案,本领域普通技术人员可以获得的所有其他实施方案,都属于本发明保护的范围。
[0026]
如上所述,通过现有的地塞米松环氧水解物的精制方法获得的8dm中均存在大于0.1%的杂质。因此,本发明的目的在于提供一种新的地塞米松环氧水解物(8dm)的精制方法,从而可以去除8dm中大于0.1%的单个杂质,提供高纯度的8dm。
[0027]
现有技术中通常采用两溶剂体系的精制方法对8dm进行提纯,经过精制后的8dm中仍存在较大量的未知的单个杂质,且杂质的结构复杂,难以确证进行结构确证,导致8dm作为原料药的起始物料进行注册申报的风险较大。发明人经试验发现,采用含有四氢呋喃或取代的四氢呋喃的三溶剂体系对8dm进行提纯处理,充分利用其它杂质与8dm在结晶溶剂中溶解度的差异,可以获得纯度达到99.5%以上,单个杂质小于0.1%,总杂质小于0.5%的8dm,并且8dm精品的收率达到90%以上。由此,本发明人完成了本发明。
[0028]
因此,在第一方面,本发明提供了一种地塞米松环氧水解物的精制方法,其特征在于,所述精制方法包括以下步骤:
[0029]
1)将地塞米松环氧水解物粗品溶于第一溶剂、第二溶剂和第三溶剂的混合物中,并升温至体系溶清得到第一体系,其中,所述第一溶剂为四氢呋喃或取代的四氢呋喃,所述第二溶剂为低级醇,所述第三溶剂为水,并且,所述第一溶剂、所述第二溶剂、所述第三溶剂与所述地塞米松环氧水解物粗品的体积质量比以ml/g计为(3-5):(3-8):(0.1-1):1;
[0030]
2)将所述第一体系蒸馏得到第一固液混合体系,作为第二体系;
[0031]
3)将所述第二体系于第一温度搅拌析晶得到第二固液混合体系,作为第三体系,其中,所述第一温度为20℃至80℃,所述搅拌析晶的时间为1小时至3小时;
[0032]
4)将所述第三体系于第二温度搅拌析晶得到第三固液混合体系,作为第四体系,其中,所述第二温度为-5℃至5℃,所述搅拌析晶的时间为3小时至5小时;
[0033]
5)将所述第四体系进行固液分离以得到固体产物。
[0034]
本发明中的8dm粗品为来自多个厂家的市售的8dm,包括西安高远生物科技有限公司(批号为210401、210502和211103)、湖南玉新药业有限公司(批号为210603)以及山东赛托生物科技有限公司(批号为53021912001)等。发明人发现通过本发明的精制方法对上述多种来源,多个批次的8dm粗品进行精制,结果均能获得单杂小于0.1%的高纯度的8dm。
[0035]
本发明的三溶剂体系是基于溶质与溶剂的相似相溶原理,小极性杂质易溶于极性相对小的呋喃类溶剂,大极性杂质易溶于极性相对大的醇类或者水中,通过调节多体系溶剂的比例达到同时除去不同极性杂质的目的。此外,发明人还在实验中发现,同一个杂质在单独的四氢呋喃和水中的溶解性与在四氢呋喃和水的混合体系的溶解性是不一样的。因此,在本发明的三溶剂体系中,所述第一溶剂、第二溶剂、第三溶剂与8dm粗品的具体体积质量比的选择十分关键,需要通过大量实验来验证。在一个实施方案中,所述第一溶剂、所述第二溶剂、所述第三溶剂与所述地塞米松环氧水解物粗品的体积质量比为(4-5):(5-6):(0.2-1):1。在一个优选实施方案中,所述体积质量比为5:5:1:1。
[0036]
本发明中首次选择使用四氢呋喃或取代的四氢呋喃作为极性相对小的溶剂用于
8dm的精制,除去8dm中小极性杂质。在一个实施方案中,所述取代的四氢呋喃可以为c1-c3烷基取代的四氢呋喃,所述取代基可位于四氢呋喃环上的任意可发生取代反应的位置。
[0037]
本发明中所述第二溶剂为低级醇,例如c1-c4醇。在一个实施方案中,所述第二溶剂可以为甲醇、乙醇、异丙醇、正丁醇或叔丁醇的一种或多种的组合。
[0038]
在步骤2)中,将从步骤1)获得的所述第一体系蒸馏得到第一固液混合体系,作为第二体系。所述蒸馏是为了除去一部分对于8dm溶解性好的溶剂,以在保证8dm的纯度的同时提高精制的收率。发明人发现如果在精制的开始就减少对8dm溶解性好的溶剂的量,则获得的8dm的精制效果差于前者。在一个实施方案中,经蒸馏得到的所述第一固液混合体系与所述地塞米松环氧水解物粗品的体积质量比以ml/g计为(2-6):1。步骤2)中的蒸馏可以通过常压蒸馏或减压蒸馏完成。在一个实施方案中,步骤2)中的所述蒸馏可以在常压下进行。
[0039]
在步骤3)中,将从步骤2)获得的所述第二体系于第一温度进行搅拌析晶。步骤3)的搅拌析晶的目的是为了充分溶解8dm中的杂质,以达到更好的除杂效果,因此第一温度为较高的温度。虽然理论上温度越高、时间越长除杂效果越好,但温度过高也会导致收率降低,时间过长则相应地提高能耗,因此发明人综合考虑精制效果和精制成本,通过多次实验选择出最合适的温度和时间。在一个实施方案中,所述第一温度为40℃至50℃,搅拌析晶的时间为1小时至3小时。在一个优选的实施方案中,搅拌析晶的时间为1小时。
[0040]
在步骤4)中,将从步骤3)获得的所述第三体系于第二温度进行搅拌析晶。步骤4)的搅拌析晶的目的是保证除杂效果的前提下提高精制收率,从而降低精制成本。在绝大部分情况下,温度越低,物质的溶解度越差,因此第二温度为较低的温度,降低温度搅拌可以使产物8dm析出,同时延长时间也可以提高精制8dm的收率。但如果第二温度过低或析晶时间过长,则会导致杂质析出,从而无法保证8dm的精制效果。在一个实施方案中,所述第一温度为-5℃至0℃,搅拌析晶的时间为3小时至5小时。在一个优选的实施方案中,搅拌析晶的时间为4小时。
[0041]
在步骤5)中,将从步骤4)获得的所述第四体系进行固液分离以得到固体产物。本领域技术人员应当理解,任何能够分离固体产物且不影响固体产物纯度和收率的固液分离方法均可以在本发明中使用。在一些实施方案中,所述精制方法还包括对步骤5)中获得的所述固体产物进行干燥。在一个实施方案中,对所述固体产物进行鼓风干燥。
[0042]
在第二方面,本发明提供了一种精制的地塞米松环氧水解物,其是通过本发明第一方面所述的精制方法制备得到的,其中所述地塞米松环氧水解物的纯度≥99.5%,并且其包含的单个杂质均小于0.1%。
[0043]
实施例
[0044]
下面结合实施例对本发明进行更为具体和详细的描述。下述实施例中的试验方法,如无特殊说明,均为常规方法。下述实施例中所用的试验材料,如无特殊说明,均为自常规化试剂商店购买所得。应注意,上文的发明内容部分以及下文的详细描述仅为具体阐释本发明之目的,无意于以任何方式对本发明进行限制。
[0045]
单溶剂精制体系
[0046]
实施例1
[0047]
将10g 8dm粗品溶于3000毫升四氢呋喃中,升温至体系溶清;将溶清后的体系在常压下蒸馏得到50毫升的固液混合体系;将所述固液混合体系于50℃搅拌析晶1小时,随后于
0℃搅拌析晶2小时;最后将得到的固液混合体系进行固液分离,收集固体产物,并对所述固体产物进行干燥,得到8.0g的8dm精品,精制收率为77.9%,经hplc检测的纯度为99.661%,最大单杂为0.103%,单溶剂精制体系的收率偏低。
[0048]
两溶剂精制体系
[0049]
实施例2
[0050]
将50g 8dm粗品溶于含有1000毫升正丁醇和10毫升水的混合溶剂中,升温至体系溶清;将溶清后的体系在常压下蒸馏得到200毫升的固液混合体系;将所述固液混合体系于50℃搅拌析晶1小时,随后于5℃搅拌析晶4小时;最后将得到的固液混合体系进行固液分离,收集固体产物,并对所述固体产物进行干燥,得到42.5g 8dm精品,精制收率为85%,经hplc检测的纯度为99.467%,含有两种大于0.1%的单质,最大单杂为0.140%,该双溶剂精制体系得到的8dm纯度偏低,含有大于0.1%的单杂。
[0051]
三溶剂精制体系
[0052]
实施例3
[0053]
将50g 8dm溶于含有250毫升丙酮、250毫升乙醇和50毫升水的混合溶剂中,升温至体系溶清;将溶清后的体系在常压下蒸馏得到200毫升的固液混合体系;将所述固液混合体系于50℃搅拌析晶1小时,随后于0℃搅拌析晶4小时;最后将得到的固液混合体系进行固液分离,收集固体产物,并对所述固体产物进行干燥,得到40g 8dm精品,精制收率为80%,经hplc检测的纯度为99.489%,含有三种大于0.1%的杂质,最大单杂为0.136%,该三溶剂精制体系的收率偏低,纯度偏低,含有大于0.1%的单杂。
[0054]
实施例4
[0055]
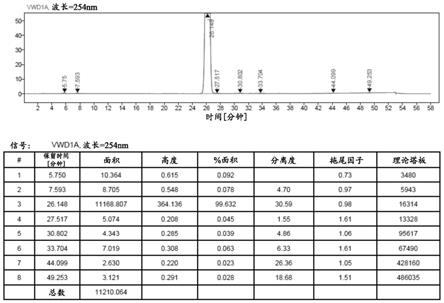
将50g 8dm溶于含有250毫升四氢呋喃、250毫升甲醇和50毫升水的混合溶剂中,升温至体系溶清;将溶清后的体系在常压下蒸馏得到200毫升的固液混合体系;将所述固液混合体系于50℃搅拌析晶1小时,随后于0℃搅拌析晶4小时;最后将固液混合体系进行固液分离,收集固体产物,并对所述固体产物进行干燥,得到47.55g高纯度的8dm精品,精制收率为95.1%,经hplc检测的纯度为99.632%,最大单杂为0.092%,如图1所示。
[0056]
实施例5
[0057]
将50g 8dm溶于含有250毫升四氢呋喃、250毫升乙醇和50毫升水的混合溶剂中,升温至体系溶清;将溶清后的体系在常压下蒸馏得到200毫升的固液混合体系;将所述固液混合体系于50℃搅拌析晶1小时,随后于0℃搅拌析晶4小时;最后将固液混合体系进行固液分离,收集固体产物,并对所述固体产物进行干燥,得到46.2g高纯度的8dm精品,精制收率为92.4%,经hplc检测的纯度为99.563%,最大单杂为0.086%,如图2所示。
[0058]
实施例6
[0059]
将50g 8dm溶于含有250毫升2-甲基四氢呋喃、300毫升正丁醇和10毫升水的混合溶剂中,升温至体系溶清;将溶清后的体系在常压下蒸馏得到200毫升的固液混合体系;将所述固液混合体系于60℃搅拌析晶1小时,随后于5℃搅拌析晶4小时;最后将固液混合体系进行固液分离,收集固体产物,并对所述固体产物进行干燥,得到46.6g高纯度的8dm精品,精制收率为93.2%。经hplc检测的纯度为99.636%,最大单杂为0.093%,如图3所示。
[0060]
以上对本发明所提供的8dm的精制方法进行了详细介绍,本文中应用了具体实施例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的
方法及其核心思想;同时,对于本领域的一般技术人员,依据本发明的思想,在具体实施方式及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本发明的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1