一种SOS1泛KRAS抑制剂手性中间体的合成方法与流程
一种sos1泛kras抑制剂手性中间体的合成方法
技术领域
1.本发明属于药物合成技术领域,涉及一种sos1泛kras抑制剂手性中间体的合成方法。
背景技术:
2.根据国家癌症中心发布的2019年全国癌症报告显示,肺癌、胃癌和结直肠癌是我国发病率排名前三位的恶性肿瘤,而kras突变发生于约25%的癌症病例中,常见于肺癌(10-20%)、胰腺癌(80-90%)和结直肠癌(40-50%)。
3.kras是前线信号传导反应靶点的一种,可触发将细胞表面连接至细胞核的一系列信号分子激活,从而控制正常细胞的生长、存活和分化。kras在一系列常见癌症的细胞信号传导中起着核心作用,加上其心形结构,所以它被称为“肿瘤跳动的心脏”。因此,阻断kras具有使许多肿瘤患者受益的巨大潜力,但是发现和开发针对该主要靶点的有效疗法极具挑战性。
4.类bi 1701963(结构如下所示)作为sos1:kras小分子抑制剂的出现,打破了多年来没有专门针对kras基因的抑制剂的困局。
[0005][0006]
临床前数据表明,kars g12c和sos1泛kras抑制剂可提高抗肿瘤活性。sos1泛kras抑制剂通过将kras平衡从激活状态转到失活状态,可能使kras g12c突变肿瘤对结合到kras(关闭状态)的共价kras g12c抑制剂更敏感。
[0007]
kras g12c突变存在于约14%的非小细胞肺腺癌(nsclc)患者,3-4%的结直肠癌患者以及其他类型癌症的分支患者中。以kras g12c突变为特征的肿瘤通常预后不佳并对治疗耐药,携带这些突变的患者的治疗选择很少。目前正在一项i/ii期临床试验中对于治疗经分子学鉴定的kras g12c阳性晚期实体瘤患者进行评价,以评估单药及与mek抑制剂联合使用的安全性、耐受性、药代动力学和药效学性质以及初步有效性。
[0008]
bi 1701963是一种可口服的在研小分子药物,可与sos1的催化结构域结合,从而防止与kras发生相互作用(关闭),并同时阻断sos1驱动的反馈。这能减少kras(开启)的形成,并因此抑制kras依赖性癌症中的mapk通路信号转导。对sos1的选择性抑制是一个治疗概念,可以不受kras突变类型的影响去阻断kras(泛kras方法)。hofmann mh等人近期在美国癌症研究协会(aacr)期刊cancer discovery中发表的论文报道,sos1:kras抑制剂对广泛kras等位基因具有活性,包括所有主要的g12d/v/c和g13d癌蛋白。目前正在一项i期临床
试验中对bi 1701963治疗晚期kras突变癌症患者进行评价,以评估bi 1701963单药及与mek抑制剂联合使用的安全性、耐受性、药代动力学和药效学性质以及初步有效性。
技术实现要素:
[0009]
本发明的目的是提供一种sos1泛kras抑制剂手性中间体的合成方法,以能够原料易得、成本低、工艺条件温和、收率和光学纯度高、后续分离简单、适于规模化生产的合成类bi 1701963合成过程中的重要中间体。
[0010]
为实现此目的,在基础的实施方案中,本发明提供一种sos1泛kras抑制剂手性中间体的合成方法,所述的合成方法包括如下步骤:
[0011]
(1)式6化合物与正丁基锂反应,经dmf淬灭,得到式7化合物,
[0012][0013]
(2)式7化合物与手性诱导试剂(s)-叔丁基亚磺酰胺经过脱水缩合反应得到式8化合物,
[0014][0015]
(3)式8化合物与甲基溴化镁反应得到式9化合物,
[0016][0017]
(4)式9化合物经盐酸气/二氧六环溶液得到式1化合物,即sos1泛kras抑制剂手性中间体,
[0018][0019]
其中:r4为chf2或cf3,r6为f或ch3。
[0020]
本发明合成的sos1泛kras抑制剂手性中间体是类bi 1701963合成过程中最重要,也是成本占比最高的中间体,它们的结构如下:
[0021][0022]
式2(r)-1-(3-(二氟甲基)-2-氟苯基)乙胺盐酸盐
[0023][0024]
式3(r)-1-(2-甲基-3-(三氟甲基)苯基)乙胺盐酸盐
[0025][0026]
式4(r)-1-(3-(二氟甲基)-2-甲基苯基)乙胺盐酸盐
[0027][0028]
式5(r)-1-(2-氟-3-(三氟甲基)苯基)乙胺盐酸盐
[0029]
现有技术中,这四个化合物的合成路线如下,都存在如下问题:
[0030][0031]
(1)通过苯乙酮化合物跟叔丁基亚磺酰胺反应,需要缩水剂钛酸异丙酯/钛酸乙酯,这类缩水剂价格比较高,而且后处理麻烦,产生大量悬浊的沉淀物,很难过滤处理,工业生产难度比较大。同时苯乙酮的原料都是用溴苯与贵重金属催化剂钯,或者需要使用有机锡试剂/格式试剂交换来实现,造成方法成本高,收率低。
[0032]
(2)席夫碱化合物手性还原过程使用三仲丁基硼氢化锂,反应需要深冷-78℃无水无氧操作,工业生产成本高,操作困难。
[0033]
(3)工艺路线总收率低,成本高,不利于工业生产。
[0034]
在一种优选的实施方案中,本发明提供一种sos1泛kras抑制剂手性中间体的合成方法,其中步骤(1)中,反应温度为-100℃~-50℃,反应时间为0.1小时~2小时。
[0035]
在一种优选的实施方案中,本发明提供一种sos1泛kras抑制剂手性中间体的合成方法,其中步骤(1)中,反应完成后将反应液倒入磷酸的水溶液中淬灭,加入甲基叔丁基醚萃取,分层,水相用甲基叔丁基醚萃取后合并所有有机相,无水硫酸钠干燥过夜,减压浓缩,得式7化合物。
[0036]
在一种优选的实施方案中,本发明提供一种sos1泛kras抑制剂手性中间体的合成方法,其中步骤(2)中,反应温度为25℃~85℃,反应时间为4小时~10小时。
[0037]
在一种优选的实施方案中,本发明提供一种sos1泛kras抑制剂手性中间体的合成
方法,其中步骤(2)中,反应完成后过滤反应液,滤饼用1,2-二氯乙烷洗涤,合并有机相,减压浓缩,得粗产物过200-300目硅胶柱,减压浓缩得式8化合物。
[0038]
在一种优选的实施方案中,本发明提供一种sos1泛kras抑制剂手性中间体的合成方法,其中步骤(3)中,反应温度为-50℃~25℃,反应时间为2小时~8小时。
[0039]
在一种优选的实施方案中,本发明提供一种sos1泛kras抑制剂手性中间体的合成方法,其中步骤(3)中,反应完成后将反应液倒入饱和氯化铵水溶液淬灭,加入甲基叔丁基醚萃取分层,水相用甲基叔丁基醚萃取,合并所有有机相,用饱和食盐水洗涤,无水硫酸钠干燥过夜,减压浓缩得式9化合物。
[0040]
在一种优选的实施方案中,本发明提供一种sos1泛kras抑制剂手性中间体的合成方法,其中步骤(4)中,反应温度为0℃~50℃,反应时间为0.5小时~2小时。
[0041]
在一种优选的实施方案中,本发明提供一种sos1泛kras抑制剂手性中间体的合成方法,其中步骤(1)、步骤(3)的反应均在氮气保护下进行。
[0042]
本发明的有益效果在于,利用本发明的合成方法,能够原料易得、成本低、工艺条件温和、收率和光学纯度高、后续分离简单、适于规模化生产的合成类bi 1701963合成过程中的重要中间体。
[0043]
本发明通过高选择性的不对称合成方法来合成类bi 1701963合成过程中的重要手性中间体,通过溴苯类原料做成苯甲醛,然后苯甲醛与手性诱导试剂叔丁基亚磺酰胺脱水缩合生成席夫碱,然后与金属有机试剂加成反应,得到高选择性的手性中间体。与现有报到的合成方法相比,本发明原料简单易得,收率和手性选择性更好,更适合工业化放大生产。
附图说明
[0044]
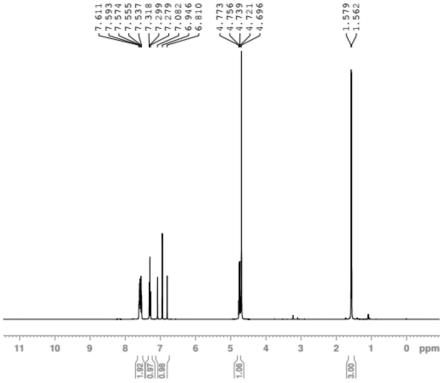
图1为实施例1制备得到的(r)-1-(3-(二氟甲基)-2-氟苯基)乙胺盐酸盐的核磁检测结果图。
[0045]
图2为实施例1制备得到的(r)-1-(3-(二氟甲基)-2-氟苯基)乙胺盐酸盐的高效液相检测结果图。
[0046]
图3为实施例2制备得到的(r)-1-(2-甲基-3-(三氟甲基)苯基)乙胺盐酸盐的核磁检测结果图。
[0047]
图4为实施例2制备得到的(r)-1-(2-甲基-3-(三氟甲基)苯基)乙胺盐酸盐的高效液相检测结果图。
[0048]
图5为实施例3制备得到的(r)-1-(3-(二氟甲基)-2-甲基苯基)乙胺盐酸盐的高效液相检测结果图。
[0049]
图6为实施例4制备得到的(r)-1-(2-氟-3-(三氟甲基)苯基)乙胺盐酸盐的核磁检测结果图。
[0050]
图7为实施例4制备得到的(r)-1-(2-氟-3-(三氟甲基)苯基)乙胺盐酸盐的高效液相检测结果图。
具体实施方式
[0051]
以下结合实施例对本发明的具体实施方式作出进一步的说明。
[0052]
实施例1:(r)-1-(3-(二氟甲基)-2-氟苯基)乙胺盐酸盐的合成与检测
[0053]
1)式10化合物与二乙胺基三氟化硫(dast)反应得到式11化合物
[0054][0055]
洗净烘干的三口烧瓶中,氮气保护,投入式10原料3-溴-2-氟苯甲醛(购于上海毕得医药)10g,溶解在50ml二氯甲烷中,冷却至0℃,缓慢滴入12g二乙胺基三氟化硫(dast),控温0℃,加完后,缓慢升温至室温,搅拌1-2小时,tlc检测反应完成。反应完成后将反应液倒入冰水中,分层,水相分别用25ml二氯甲烷再萃取两次,合并所有有机相,无水硫酸钠干燥过夜,减压浓缩,得粗产品化合物14.5g,过200-300目硅胶柱,减压浓缩得式11化合物9.2g,纯度95%,收率83%。
[0056]
2)式11化合物与正丁基锂反应,经二甲基甲酰胺(dmf)淬灭,得式12化合物醛
[0057][0058]
洗净烘干的三口烧瓶中,氮气保护,投入式11化合物5g,加入四氢呋喃50ml,降温至-78℃,缓慢滴入2.5n正丁基锂10.5ml,控制-78℃搅拌1小时,检测反应完成,-78℃下缓慢滴加5g dmf,搅拌反应30分钟,检测反应完成。反应完成后将反应液倒入稀磷酸的水溶液中淬灭,加入50ml甲基叔丁基醚萃取,分层,水相分别用50ml甲基叔丁基醚萃取两次,合并所有有机相,用50ml饱和食盐水洗一次,无水硫酸钠干燥过夜,减压浓缩得式12化合物4.7g,不用做进一步纯化,可直接进行下步反应。
[0059]
3)式12化合物与手性诱导试剂(s)-叔丁基亚磺酰胺经过脱水缩合反应得到式13化合物
[0060][0061]
洗净烘干的三口烧瓶中,加入式12化合物4.7g、1,2-二氯乙烷(dce)23.5ml、无水硫酸铜(cuso4)10.5g、4-甲基苯磺酸吡啶(ppts)260mg、s-叔丁基亚磺酰胺3.9g,缓慢升温至50℃,搅拌反应6-8小时,tlc检测反应完成。过滤反应液,滤饼用1,2-二氯乙烷(dce)洗1-2次,合并有机相,减压浓缩,得粗品化合物7.8g,过200-300目硅胶柱,减压浓缩得式13化合物4.3g。
[0062]
4)式13化合物与甲基溴化镁反应得到式14化合物
[0063][0064]
洗净烘干的三口烧瓶中,氮气保护,加入式13化合物4.0g、二氯甲烷(dcm)20ml,降温至-30℃,缓慢滴加3n的甲基溴化镁溶液7.0ml,滴加完成后缓慢升温至0℃,保温反应3-4小时,tlc检测反应完成。反应液倒入饱和氯化铵水溶液淬灭,加入20ml甲基叔丁基醚萃取分层,水相分别用10ml甲基叔丁基醚萃取2次,合并所有有机相,用25ml饱和食盐水洗1次,无水硫酸钠干燥过夜,减压浓缩,得5.5g粗品化合物,过200-300目硅胶柱,减压浓缩得式14化合物2.5g,收率59.1%(hplc》97%de》98%)。
[0065]
5)式14化合物经盐酸气/二氧六环溶液得到式2化合物
[0066][0067]
洗净烘干的三口烧瓶中,加入式14化合物2.5g、4n的盐酸气/二氧六环溶液25ml,搅拌反应1小时,析出白色固体,hplc检测反应完成。过滤,烘干得式2化合物1.7g,收率89%,五步反应总收率为30.4%,其中后四步反应的总收率为36.6%(hplc》98%ee》98%)。h-nmr(400mhz,d2o)δ:7.537-7.611(m,2h),7.279-7.318(m,1h),6.810-7.082(m,1h),4.696-4.773(m,1h),1.562-1.579(d,3h);lcms(m/z,190.1,m+1);化学纯度hplc=98.9%(agilent 1100,ymc ods-aq 3um 120a 4.6
×
50mm,210nm,acn/h2o/缓冲液)。其核磁氢谱及高效液相色谱检测谱图见图1-2。
[0068]
实施例2:(r)-1-(2-甲基-3-(三氟甲基)苯基)乙胺盐酸盐的合成与检测
[0069]
1)式16化合物与正丁基锂反应,经dmf淬灭,得到式17化合物
[0070][0071]
洗净烘干的三口烧瓶中,氮气保护,加入式16化合物(购于上海毕得医药)20g,加入四氢呋喃200ml,降温至-78℃,缓慢滴加2.5n正丁基锂36ml,控温-78℃搅拌1小时,检测反应完成。-78℃下缓慢滴加20g dmf,搅拌反应30分钟,检测反应完成。反应液倒入稀磷酸水溶液中淬灭,加入200ml甲基叔丁基醚萃取分层,水相分别用100ml甲基叔丁基醚萃取2次,合并有机相,用200ml饱和食盐水洗一次,无水硫酸钠干燥过夜,减压浓缩得式17化合物15g,不用做进一步纯化,可直接进行下步反应。
[0072]
2)式17化合物与手性诱导试剂(s)-叔丁基亚磺酰胺经过脱水缩合反应得到式18化合物
[0073][0074]
洗净烘干的三口烧瓶中,加入式17化合物5g,加入1,2二氯乙烷(dce)100ml,加入无水硫酸铜(cuso4)8.5g,加入125mg ppts,加入s-叔丁基亚磺酰胺3.8g,加热至50℃,搅拌反应6-8小时,tlc检测反应完成。过滤,滤饼用1,2-二氯乙烷(dce)洗1-2次,合并有机相减压浓缩得粗品化合物8.2g,过200-300目硅胶柱,减压浓缩得式18化合物6.1g。
[0075]
3)式18化合物与甲基溴化镁反应得到式19化合物
[0076][0077]
洗净烘干的三口烧瓶中,氮气保护,加入式18化合物32.8g,加入二氯甲烷(dcm)165ml,降温至-30℃,缓慢滴加3n的甲基溴化镁溶液57ml,滴加完成后缓慢升温至0℃,保温反应3-4小时,tlc检测反应完成。反应液倒入饱和氯化铵水溶液淬灭,加入165ml甲基叔丁基醚萃取分层,水相分别用90ml甲基叔丁基醚萃取2次,合并有机相,用200ml饱和食盐水洗1次,无水硫酸钠干燥过夜,减压浓缩得35g粗品化合物,过200-300目硅胶柱得式19化合物21g,收率60.7%(hplc》97%de》98%)。
[0078]
4)式19化合物经盐酸气/二氧六环溶液得到式3化合物
[0079][0080]
洗净烘干的三口烧瓶中,加入式19化合物21g,加入4n的盐酸气/二氧六环溶液210ml,搅拌反应1小时,析出白色固体,hplc检测反应完成。过滤,烘干得式3化合物15.0g,收率92%,四步反应总收率41.8%(hplc》98%ee》98%)。h-nmr(600mhz,dmso-d6)δ8.60(s,3h),7.93(d,1h),7.71(dd,1h),7.52(t,1h),4.72(q,1h),2.45(d,3h),1.51(d,3h);lcms(m/z,204.3,m+1);化学纯度hplc=98.6%(agilent 1100,ymc ods-aq 3um 120a 4.6
×
50mm,210nm,acn/h2o/缓冲液)。其核磁氢谱及高效液相色谱检测谱图见图3-4。
[0081]
实施例3:(r)-1-(3-(二氟甲基)-2-甲基苯基)乙胺盐酸盐的合成与检测
[0082]
1)式21化合物与二乙胺基三氟化硫(dast)反应得到式22化合物
[0083]
[0084]
洗净烘干的三口烧瓶中,氮气保护,投入式21原料2-甲基-3-溴-苯甲醛(购于上海毕得医药)20g,溶解在200ml二氯甲烷中,冷却至0℃,缓慢滴入32.3g二乙胺基三氟化硫(dast),控温0℃,加完后,缓慢升温至室温,搅拌1-2小时,tlc检测反应完成。反应完成后将反应液倒入冰水中,分层,水相分别用100ml二氯甲烷再萃取两次,合并有机相,无水硫酸钠干燥过夜,减压浓缩,得粗产品化合物22g。过200-300目硅胶柱,减压浓缩得式22化合物19g,收率85.5%(hplc》95%)。
[0085]
2)式22化合物与正丁基锂反应,经dmf淬灭,得式23化合物
[0086][0087]
洗净烘干的三口烧瓶中,氮气保护,加入式22化合物19g,加入四氢呋喃190ml,降温至-78℃,缓慢滴加2.5n正丁基锂41.5ml,控温-78℃搅拌1小时,检测反应完成,-78℃下缓慢滴加19g dmf,搅拌反应30分钟,检测反应完成。反应液倒入稀磷酸水溶液中淬灭,加入200ml甲基叔丁基醚萃取分层,水相分别用100ml甲基叔丁基醚萃取2次,合并有机相,用200ml饱和食盐水洗一次,无水硫酸钠干燥过夜,减压浓缩得式23化合物14.7g,不用做进一步纯化,可直接进行下步反应。
[0088]
3)式23化合物与手性诱导试剂(s)-叔丁基亚磺酰胺经过脱水缩合反应得到式24化合物
[0089][0090]
洗净烘干的三口烧瓶中,加入式23化合物5g,加入1,2二氯乙烷(dce)50ml,加入无水硫酸铜(cuso4)9.5g,加入125mg ppts,加入s-叔丁基亚磺酰胺4.3g,加热至50℃,搅拌反应6-8小时,tlc检测反应完成。过滤,滤饼用1,2-二氯乙烷(dce)洗1-2次,合并有机相减压浓缩得粗品化合物8.5g,过200-300目硅胶柱,减压浓缩得式24化合物6.2g。
[0091]
4)式24化合物与甲基溴化镁反应得到式25化合物
[0092][0093]
洗净烘干的三口烧瓶中,氮气保护,加入式24化合物27g,加入二氯甲烷(dcm)135ml,降温至-30℃,缓慢滴加3n的甲基溴化镁溶液50ml,滴加完成后缓慢升温至0℃,保温反应3-4小时,tlc检测反应完成。反应液倒入饱和氯化铵水溶液淬灭,加入135ml甲基叔丁基醚萃取分层,水相分别用70ml甲基叔丁基醚萃取2次,合并有机相,用150ml饱和食盐水洗
1次,无水硫酸钠干燥过夜,减压浓缩得30g粗品化合物,过200-300目硅胶柱得式25化合物18.5g,收率64.7%(hplc》97%de》98%)。
[0094]
5)式25化合物经盐酸气/二氧六环溶液得到式4化合物
[0095][0096]
洗净烘干的三口烧瓶中,加入式25化合物18.5g,加入4n的盐酸气/二氧六环溶液185ml,搅拌反应1小时,析出白色固体,hplc检测反应完成。过滤,烘干得式4化合物12.5g,收率88.6%,五步反应总收率为37.9%,其中后四步反应总收率为44.3%(hplc》98%ee》98%)。lcms(m/z,186.4,m+1);化学纯度hplc=98.5%(agilent 1100,ymc ods-aq 3um 120a 4.6
×
50mm,210nm,acn/h2o/缓冲液),其高效液相谱图见图5。
[0097]
实施例4:(r)-1-(2-氟-3-(三氟甲基)苯基)乙胺盐酸盐的合成与检测
[0098]
1)式27化合物与正丁基锂反应,经dmf淬灭,得式28化合物
[0099][0100]
洗净烘干的三口烧瓶中,氮气保护,加入式27化合物(购于上海毕得医药)25g,加入四氢呋喃250ml,降温至-78℃,缓慢滴加2.5n正丁基锂53ml,控温-78℃搅拌1小时,检测反应完成,-78℃下缓慢滴加25g dmf,搅拌反应30分钟,检测反应完成。反应液倒入稀磷酸水溶液中淬灭,加入250ml甲基叔丁基醚萃取分层,水相分别用100ml甲基叔丁基醚萃取2次,合并有机相,用250ml饱和食盐水洗一次,无水硫酸钠干燥过夜,减压浓缩得式28化合物19.5g,不用做进一步纯化,可直接进行下步反应。
[0101]
2)式28化合物与手性诱导试剂(s)-叔丁基亚磺酰胺经过脱水缩合反应得到式29化合物
[0102][0103]
洗净烘干的三口烧瓶中,加入式28化合物19.5g,加入1,2-二氯乙烷(dce)200ml,加入无水硫酸铜(cuso4)32.5g,加入487mg ppts,加入s-叔丁基亚磺酰胺14.7g,缓慢升温至50℃,搅拌反应6-8小时,tlc检测反应完成。过滤,滤饼用1,2-二氯乙烷(dce)洗1-2次,合并有机相减压浓缩得粗品化合物32g,过200-300目硅胶柱,减压浓缩得式29化合物22.6g。
[0104]
3)式29化合物与甲基溴化镁反应得到式30化合物
[0105][0106]
洗净烘干的三口烧瓶中,氮气保护,加入式29化合物13.2g,加入二氯甲烷(dcm)135ml,降温至-30℃,缓慢滴加3n的甲基溴化镁溶液22.4ml,滴加完成后缓慢升温至0℃,保温反应3-4小时,tlc检测反应完成。反应液倒入饱和氯化铵水溶液淬灭,加入135ml甲基叔丁基醚萃取分层,水相分别用90ml甲基叔丁基醚萃取2次,合并有机相,用100ml饱和食盐水洗1次,无水硫酸钠干燥过夜,减压浓缩得18g粗品化合物,过200-300目硅胶柱得式30化合物8.4g,收率60.4%(hplc》97%de》98%)。
[0107]
4)式30化合物经盐酸气/二氧六环溶液得到式5化合物
[0108][0109]
洗净烘干的三口烧瓶中,加入式30化合物8.4g,加入4n的盐酸气/二氧六环溶液84ml,搅拌反应1小时,析出白色固体,hplc检测反应完成。过滤,烘干得式1化合物5.9g,收率89.9%,四步反应总收率为40.2%(hplc》98%ee》98%)。h-nmr(400mhz,d2o)δ:7.68-7.76(m,2h),7.36-7.41(m,1h),4.68-4.84(m,1h),1.62-1.64(d,3h);lcms(m/z,208.3,m+1);化学纯度hplc=99.5%(agilent 1100,ymc ods-aq 3um 120a 4.6
×
50mm,210nm,acn/h2o/缓冲液)。其核磁氢谱及高效液相色谱检测谱图见图6-7。
[0110]
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若对本发明的这些修改和变型属于本发明权利要求及其同等技术的范围之内,则本发明也意图包含这些改动和变型在内。上述实施例或实施方式只是对本发明的举例说明,本发明也可以以其它的特定方式或其它的特定形式实施,而不偏离本发明的要旨或本质特征。因此,描述的实施方式从任何方面来看均应视为说明性而非限定性的。本发明的范围应由附加的权利要求说明,任何与权利要求的意图和范围等效的变化也应包含在本发明的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1