一种通过基因组编辑创制番茄雄性不育材料的方法及应用
1.本发明属于生物技术领域,具体涉及一种通过基因组编辑创制番茄雄性不育材料的方法及应用。
背景技术:
2.番茄(solanum lycopersicum)是全球最重要的园艺经济作物之一,也是我国最主要的消费蔬菜。番茄属于严格的闭花授粉作物,具有明显的杂种优势,对其利用能极大提高番茄的产量、抗病及抗逆表现,是重要的育种手段。目前番茄杂交种生产仍然以费时、费力、制种成本高、种子纯度无保障的人工去雄授粉法为主,在制种时引入雄性不育系可以优化制种程序,降低劳动力成本,提高杂种种子纯度以及避免亲本流失。因此开发更便于生产应用的雄性不育系是番茄杂交育种研究的重点。
3.植物中许多重要的转录因子家族成员参与调控雄蕊及花药的发育,突变后往往会造成植物雄性不育,比如拟南芥(arabidopsis thaliana)dyt1(dys-functional tapetum1)基因及其在水稻中的直系同源基因udt1(undeveloped tapetum1)均属于bhlh转录因子家族,调控绒毡层的早期发育,突变将导致绒毡层发育不正常从而引起雄性不育(fuet al.,2014)。拟南芥中apelata3(ap3)是花发育b类功能基因,是mads-box基因家族成员,该基因发生突变将导致花器官发生同源异型突变,原本的花瓣转变成萼片状器官,同时原本的雄蕊也转变成雌蕊状结构,即雄性不育。番茄雄性不育系通常是将突变体材料中的不育基因经反复回交转育到所需要的亲本材料中形成新的不育系,由于不育基因常与部分不良基因连锁,用传统的回交转育方法培育优良的不育系耗时费力,极大地限制了番茄品种的更新迭代速率。
4.基因组编辑技术是一种可在基因组水平上对dna序列进行改造的分子生物学技术,其原理是利用核酸内切酶切断目的dna序列,通过dna修复过程中产生的一系列突变达到定向改造基因组的目的。其中,crispr-cas(clustered regularly interspaced short palindromic repeats,crispr-crispr associated protein)系统是一类来源于细菌适应性免疫防御系统的核酸酶,属于第三代基因编辑工具。该系统仅需要一个包含dna结合及切割功能的核酸酶蛋白和一条用来指导核酸酶蛋白与dna结合的向导rna即可实现对核心亲本目标性状的快速、精准改良,且避免了杂交育种中的连锁累赘,在作物定向育种中具有巨大的应用价值。
技术实现要素:
5.本发明的目的在于克服现有技术的不足,提供一种通过基因组编辑创制番茄雄性不育材料的方法及应用。
6.本发明的目的是通过以下技术方案来实现的:
7.一种通过基因组编辑创制番茄雄性不育材料的方法,包括以下步骤:
8.利用crispr/cas9技术对solyc02g084630基因进行编辑,使solyc02g084630基因
功能丧失,得到番茄雄性不育材料,所述solyc02g084630基因的序列如seq id no.1所示。
9.进一步的,所述crispr/cas9技术中的grna靶位点选择在solyc02g084630基因的第一外显子区域,选取原型间隔序列毗邻基序(protospacer-associated motif,pam)上游19-20bp碱基长度,即5'-n19-20ngg-3',ngg为pam序列,n19-20代表19-20bp碱基的识别序列,所述solyc02g084630基因的第一外显子区域的序列如seq id no.2所示。
10.进一步的,所述grna靶位点的序列包括target1和/或target2,所述target1、target2序列如下:
11.target1:5
’‑
tgaaaactcgacaaacaggc-3’;
12.target2:5
’‑
tcatcatgctatcaagcacc-3’。为保证编辑效率,可以选择两个靶位点。
13.进一步的,利用crispr/cas9技术对solyc02g084630基因进行编辑使solyc02g084630基因功能丧失的方法为:利用crispr/cas9技术使solyc02g084630基因形成dna双链断裂位点(double-strand breaks,dbss),利用植物自身对dbs的修复机制,在断裂位点引入插入、缺失或替换,导致solyc02g084630基因的功能丧失。
14.进一步的,所述crispr/cas9技术对solyc02g084630基因进行编辑的方法步骤包括:
15.s1、根据识别序列设计oligo序列引物,所述引物包括target1-f和/或target2-f,
16.所述target1-f为5
’‑
atatatggtctcgattg n19-20 gttttagagctagaaatag-3’,
17.所述target2-f为5
’‑
attattggtctcgaaac n19-20caatctcttagtcgactct-3’,其中,n19-20代表19-20bp碱基的识别序列;
18.s2、以中间载体pcbc-dt1t2为模板,加入所述oligo序列引物,用高保真酶进行pcr扩增,回收pcr产物;
19.s3、将pkse401载体及所述pcr产物用bsai内切酶进行酶切,酶切产物回收后,按照载体:片段=1:7(摩尔比)进行混合,通过t4 dna连接酶进行连接反应;
20.s4、将连接后的载体转入大肠杆菌感受态,挑取单克隆进行菌体pcr,菌体pcr所用引物序列如seq id no.3、seq id no.4所示;
21.s5、将菌体pcr结果鉴定为阳性的质粒进行测序分析,测序所用引物序列如seq id no.3所示,将测序正确的质粒,转入农杆菌gv3101感受态,并进行菌体pcr,得到包含正确质粒的阳性农杆菌;
22.s6、以番茄子叶为外植体,通过叶盘转化法进行阳性农杆菌gv3101介导的遗传转化,经过卡那霉素抗性筛选,获得再生植株。
23.进一步的,所述crispr/cas9技术对solyc02g084630基因进行编辑的方法还包括:
24.s7、根据cas9序列设计引物,提取再生植株基因组dna,并以此为模板进行pcr扩增,进行转基因阳性株系的检测;
25.s8、根据靶位点上下游序列设计引物,以转基因阳性株系基因组dna为模板进行pcr扩增,对扩增产物进行测序,对转基因阳性株系是否发生基因组编辑,以及编辑类型进行测序分析;
26.s9、对测序分析中得到的阳性编辑植株进行花器官表型鉴定,同时对花粉活性进行检测,从而获得solyc02g084630基因经编辑后的雄性不育新种质。
27.本发明提供一种获得番茄雄性不育系的方法,通过上述方法得到的不育材料与目
标材料进行杂交和回交,从而使目标材料获得番茄雄性不育的性状和基因突变。
28.本发明还提供一种上述的方法获得的番茄雄性不育系在杂交育种和制种中的应用。
29.进一步的所述的杂交育种和制种是指将所述番茄雄性不育系作为母本与其他父本进行杂交。还包括将获得的所述番茄雄性不育系与其他目标材料进行杂交和回交,从而使目标材料获得番茄雄性不育的性状和基因突变。
30.本发明的有益效果是:
31.本发明利用crispr/cas9技术对solyc02g084630基因进行编辑,使solyc02g084630基因功能丧失,得到番茄雄性不育材料,这些番茄雄性不育材料育性稳定,可以创制不同遗传背景的雄性不育系,从而可以应用于杂交育种和制种。
附图说明
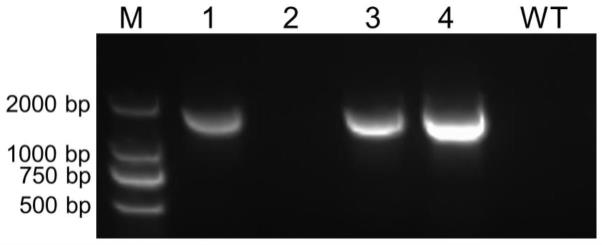
32.图1为琼脂糖凝胶电泳图;
33.图2为基因编辑部位碱基序列的测序峰图;
34.图3为花朵对比图和花粉染色图。
具体实施方式
35.下面结合附图进一步详细描述本发明的技术方案,但本发明的保护范围不局限于以下所述。
36.实验材料
37.本研究所使用的番茄材料t048,来自课题组已收集番茄品种中分离的优良高代自交系。用于载体构建的大肠杆菌(escherichia coli)dh5α感受态及用于遗传转化的根癌农杆菌(agrobacterium tumefaciens)gv3101感受态,均购自北京擎科生物科技有限公司。研究使用的crispr/cas9系统相关载体pkse401购自addgene载体库(https://www.addgene.org/)。easytaq dna聚合酶购自北京全式金生物技术有限公司,primestar hs dna聚合酶购自大连takara公司;限制性内切酶及t4 dna连接酶购自neb公司。
38.相关培养基配置如下:
39.种子萌发培养基:ms基础盐2.2g/l,r3维生素(硫胺素1g/l;烟酸0.5g/l;吡哆醇0.5g/l)50μl/l,琼脂粉8g/l,调ph至5.9,121℃高压灭菌20min。
40.预培养及共培养培养基(kcm固体):ms基础盐4.4g/l,蔗糖20g/l,磷酸二氢钾(kh2po4)200mg/l,琼脂粉8g/l,硫胺素0.9mg/l,乙酰丁香酮200μm,2,4-d 0.2mg/l,激动素0.1mg/l,调ph至5.8,121℃高压灭菌20min。
41.农杆菌侵染培养基(kcms液体):ms基础盐4.4g/l,蔗糖20g/l,磷酸二氢钾(kh2po4)200mg/l,硫胺素0.9mg/l,乙酰丁香酮200μm,调ph至5.7,121℃高压灭菌20min。
42.筛选培养基(2z或1z):ms基础盐4.4g/l、蔗糖30g/l、琼脂8g/l、vit nitsch(1000
×
)1ml、玉米素核糖苷2mg/l或1mg/l、卡那霉素100mg/l、augmentin 100mg/l、timentin 100mg/l,调ph至5.8,121℃高压灭菌20min。
43.生根培养基(enr):ms基础盐2.2g/l、蔗糖10g/l、琼脂8g/l、vit nitsch(1000
×
)1ml、卡那霉素75mg/l、augmentin 100mg/l、timentin 100mg/l,调ph至5.8,121℃高压灭菌
20min。
44.lb培养基:胰蛋白胨10g/l、酵母提取物5g/l,氯化钠10g/l,利福平25mg/l,卡那霉素100mg/l,固体需加入15%琼脂粉,121℃高压灭菌20min。
45.实施例1靶点及引物设计
46.首先使用植物基因组编辑靶位点在线设计软件crispr-p v2.0(http://crispr.hzau.edu.cn/crispr2/)对番茄育性基因tm6(solyc02g084630)进行靶位点设计。tm6基因包含7个外显子,选择位于第一外显子区域内的若干靶位点,并对选择的靶位点进行得分及脱靶分析。为保证编辑效率,最终选择两个靶位点作为对tm6基因进行编辑的sgrna靶序列。两个靶位点sgrna序列分别为:
47.target1(seq id no.5):5
’‑
tgaaaactcgacaaacaggc-3’48.target2(seq id no.6):5
’‑
tcatcatgctatcaagcacc-3’49.根据靶位点sgrna序列进行引物设计并合成,序列如下:
50.tm6-target1-f(seq id no.7):
[0051]5’‑
atatatggtctcgattgtgaaaactcgacaaacaggcgttttagagctagaaatag-3’[0052]
tm6-target2-r(seq id no.8):
[0053]5’‑
attattggtctcgaaacggtgcttgatagcatgatgacaatctcttagtcgactct-3’[0054]
实施例2crsipr/cas9重组载体构建及农杆菌转化
[0055]
首先,以pcbc-dt1t2中间载体为模板,用tm6-target1-f及tm6-target2-r引物对进行高保真pcr扩增,pcr反应条件为:95℃预变性2min,95℃变性10sec,48℃退火10sec,72℃延伸10sec,共35个循环,72℃延伸5min,对扩增产物回收后,进行bsa i酶切;随后,将酶切产物连接到同样进行bsa i酶切的pkse401载体中,并进行大肠杆菌的转化;最后,挑取阳性克隆进行一代测序验证。将构建成功的载体命名为pkse401-tm6,并通过热激法转入农杆菌gv3101,选取阳性克隆摇菌后,加入85%甘油于-80℃冰箱中保存备用。
[0056]
实施例3番茄遗传转化
[0057]
播种消毒:番茄种子用75%酒精处理20sec,倒掉酒精,用无菌水冲洗一次,然后用5%次氯酸钠浸泡20min,之后用无菌水冲洗4次,将种子上多余水分用无菌滤纸吸干,放入种子萌发培养基,置于25℃、16h光照/8h黑暗的恒温培养室培养;
[0058]
外植体制备及预培养:待种子萌发8天后,在无菌条件下用锋利的刀片将子叶切成方块状,并转移至预培养培养基中,于25℃黑暗条件下培养1天后,可用于番茄转化。
[0059]
外植体与农杆菌共培养:将过夜活化的阳性农杆菌菌液离心,用kcms液体培养基进行菌液重悬至od600=0.1后,吸取菌液加入至预培养1天的子叶中。农杆菌侵染子叶20分钟之后,将多余菌液菌吸出,移至黑暗处共培养2天。
[0060]
抗性筛选:将共培养的子叶转至2z培养基上,每皿20-30片子叶,以保证子叶所需生长空间。双层封口膜封口后,置于25℃,16h光照/8h黑暗的恒温培养室培养两周。两周后,将子叶转移到新鲜2z培养基平板。然后每2-3周继代培养一次。2-3周后会长出白色愈伤组织。4-6周之内会出现绿芽。当芽开始出现后,每2周继代一次外植体,培养基为1z选择性培养基。
[0061]
生根培养:当幼芽长至2cm左右时,用无菌刀片将其切下置于生根培养基中。2周左右开始出现根。当植株生长足够大时,可将幼苗移栽进含蛭石和土壤混合物的小塑料盆中,
并用营养液浇灌。
[0062]
实施例4再生植株转基因阳性鉴定及基因组编辑检测
[0063]
(1)再生植株转基因阳性鉴定:分别提取再生植株及野生型t048材料的叶片基因组dna,用cas9-f和cas9-r组成的引物对进行pcr扩增,pcr反应条件为:94℃预变性3min,94℃变性30sec,53℃退火30sec,72℃延伸1min30sec,共35个循环,72℃延伸5min。cas9-f和cas9-r引物序列如下:
[0064]
cas9-f(seq id no.9):5
’‑
gcagctctccaaggacacat-3’[0065]
cas9-r(seq id no.10):5
’‑
cgtgagttcttctggccctt-3’[0066]
将上述pcr扩增产物进行1%琼脂糖凝胶电泳。结果如图1所示。从图1中可以看出:以野生型番茄的基因组dna为模板的pcr扩增产物在琼脂糖凝胶电泳检测中无特异条带,而以t0代转基因番茄(1,3,4)的基因组dna为模板的pcr扩增产物在琼脂糖凝胶电泳检测中显示一条大小为1541bp的特异条带,说明t0代转基因番茄含有cas9转基因片段。
[0067]
(2)基因组编辑检测:
[0068]
根据tm6两个靶位点附近的基因组序列设计检测引物对tm6-edit12-f及tm6-edit12-r,用于检测target1及target2靶点编辑情况。tm6-edit12-f及tm6-edit12-r引物序列如下:
[0069]
tm6-edit12-f(seq id no.11):5
’‑
gtgcagttgctacccttcaa-3’[0070]
tm6-edit12-r(seq id no.12):5
’‑
ctccaagtgcactctgatactga-3’[0071]
以上一步得到的转基因阳性株系1、3、4的基因组dna为模板进行高保真pcr扩增,pcr反应条件为:95℃预变性2min,95℃变性10sec,55℃退火10sec,72℃延伸10sec,共35个循环,72℃延伸5min。随后用tm6-edit12-f引物进行测序分析,检测基因编辑部位碱基序列的变异情况。检测结果证实,1号和4号再生植株与野生型相比,在基因编辑位点target2处均发生了序列的变异(图2),但1号植株的变异为杂合类型,双链dna其中一条发生了13bp碱基的缺失;4号植株的变异类型为纯合变异,dna双链均在同一位点处缺失了7bp碱基序列;3号植株为野生型。
[0072]
实施例4田间表型及花粉活性检测
[0073]
对突变体tm6-4植株进行育性表型鉴定,同时以野生型番茄t048作为对照。结果如图3所示,图3a和图3c为野生型番茄t048的花、花粉活性,图3b和图3d为突变体tm6-4株系的花、花粉活性。与野生型番茄t048相比,经基因组编辑产生的突变体tm6-4株系表现为柱头外露、雄蕊发育不全且花粉完全无活性。
[0074]
以上所述仅是本发明的优选实施方式,应当理解本发明并非局限于本文所披露的形式,不应看作是对其他实施例的排除,而可用于各种其他组合、修改和环境,并能够在本文所述构想范围内,通过上述教导或相关领域的技术或知识进行改动。而本领域人员所进行的改动和变化不脱离本发明的精神和范围,则都应在本发明所附权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1