过氧化碳酰胺、注射用过氧化碳酰胺制剂及其制备方法与流程
1.本发明涉及医药领域,尤其涉及一种过氧化碳酰胺、注射用过氧化碳酰胺制剂及其制备方法。
背景技术:
2.过氧化碳酰胺,也叫过氧化尿素,是一种药物,用于各种低氧血症以及急性缺氧引起的胎儿窘迫。其分子式为分子式co(nh2)2·
h2o2。
3.然而,受限于其成分本身的不稳定性以及生产过程中不可避免的引入杂质,导致现有技术制备得到的产品,其长期有效性和纯度不能够得到保证,使得其注射用制剂难以长期保存且金属杂质及其它不挥发性杂质难以去除,容易出现失效、炸瓶等问题。
技术实现要素:
4.本发明的目的在于提供一种过氧化碳酰胺、注射用过氧化碳酰胺制剂及其制备方法,以解决上述问题。
5.为了实现本发明的上述目的,特采用以下技术方案:
6.一种过氧化碳酰胺的制备方法,包括:
7.将包括过氧化氢溶液、依地酸二钠、枸橼酸、尿素在内的原料混合,反应后进行析晶处理、过滤、干燥得到过氧化碳酰胺;
8.所述枸橼酸的用量小于等于所述尿素的质量的4%。
9.优选地,所述混合包括:
10.将所述依地酸二钠加入所述过氧化氢溶液,然后再加入所述枸橼酸得到第一料液,将所述尿素加入所述第一料液中,得到混合体系;
11.所述依地酸二钠的用量为所述尿素的质量的0.1%~1.0%;
12.优选地,所述过氧化氢溶液与所述尿素的投料摩尔比为1:1~1.1:1。
13.优选地,所述反应包括:将所述混合体系在30~40℃条件下保温 5~30min,得到第二料液。
14.优选地,所述析晶处理包括:将体系先冷却至10℃,然后再冷却至-5℃,维持体系在-7℃至0℃搅拌析晶10~12h;
15.优选地,所述析晶处理之前包括:将体系使用0.45μm聚丙烯滤膜进行过滤。
16.优选地,所述过滤包括:将体系用200~400目尼龙筛布过滤,得到过氧化碳酰胺湿品。
17.优选地,所述干燥采用真空干燥,温度为30~40℃,时间为24~36h,真空度大于等于0.09mpa。
18.本技术还提供一种过氧化碳酰胺,使用所述的过氧化碳酰胺的制备方法制得。
19.本技术还提供一种注射用过氧化碳酰胺制剂的制备方法,包括:
20.将所述的过氧化碳酰胺用注射用水溶解,然后过滤除菌、冻干得到所述注射用过
氧化碳酰胺制剂。
21.优选地,所述注射用水的温度为30~40℃;
22.优选地,所述过滤除菌包括:
23.依次用0.45μm聚醚砜筒式过滤器减菌过滤、用0.22μm聚醚砜筒式过滤器进行两次除菌过滤;
24.优选地,第一次用0.22μm聚醚砜筒式过滤器进行除菌过滤在c级环境局部a级条件下进行;第二次用0.22μm聚醚砜筒式过滤器进行除菌过滤在 b级环境局部a级条件下进行;
25.优选地,所述冻干包括:在-40~-50℃条件下预冻干1~5h,然后在3h 内升温至1℃,保温6~8h;再升温至15℃,保温2~4h;再升温至22℃,保温2~4h。
26.本技术还提供一种注射用过氧化碳酰胺制剂,包括使用所述的注射用过氧化碳酰胺制剂的制备方法制得。
27.本发明的有益效果:
28.本技术提供的过氧化碳酰胺的制备方法,将包括过氧化氢溶液、依地酸二钠、枸橼酸、尿素在内的原料混合,反应后进行析晶处理、过滤、干燥得到过氧化碳酰胺;通过枸橼酸和依地酸二钠的配合使用,实现抑制过氧化氢分解,提升过氧化碳酰胺稳定性的目的;通过控制枸橼酸的用量小于等于尿素的质量的4%,使得最终所得的过氧化碳酰胺中的枸橼酸的含量维持在较低水平,从而保证了过氧化碳酰胺的安全性;通过析晶处理、过滤工序实现对杂质的去除,保证了产品的纯度;
29.本技术提供的过氧化碳酰胺及注射用过氧化碳酰胺制剂,稳定性好、纯度高、安全性好。
附图说明
30.为了更清楚地说明本技术实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本技术的某些实施例,因此不应被看作是对本技术范围的限定。
31.图1为多次给药实验兔的血管刺激性反应试验结果。
32.图2为实验兔体外红细胞的溶血性反应试验结果。
33.图3为大鼠被动皮肤过敏反应试验结果。
具体实施方式
34.需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将结合具体实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。
35.首先,对本发明进行整体性解释,具体如下:
36.一种过氧化碳酰胺的制备方法,包括:
37.将包括过氧化氢溶液、依地酸二钠、枸橼酸、尿素在内的原料混合,反应后进行析晶处理、过滤、干燥得到过氧化碳酰胺;
38.在一个可选的实施方式中,枸橼酸的用量小于等于尿素的质量的4%。
39.可选的,枸橼酸的用量可以为尿素的质量的0.1%、0.5%、1.0%、1.37%、 1.5%、
2%、2.5%、3.0%、3.5%以及4%之间的任意值。
40.过氧化碳酰胺是过氧化氢与尿素以氢键结合形成的配合物,结合力较弱,过氧化氢分子相对独立,故凡能使过氧化氢分解的因素,如:微量金属离子、光照、受热等,都会影响过氧化碳酰胺的稳定性。因此,在反应体系中加入依地酸二钠可以络合金属离子,防止过氧化氢分解。
41.枸橼酸的添加量直接影响了产物的稳定性和安全性,因为过氧化氢是一种极弱酸(ka=2.4
×
10-12
),其存在如下离解平衡:
[0042][0043]
其在中性或弱酸性条件下,离解平衡向左移,比较稳定;在碱性条件下,离解平衡向右移,开始分解。故ph值也会影响过氧化碳酰胺的稳定性。加入一定量的枸橼酸来调节ph值,以抑制过氧化氢的离解。枸橼酸添加量小于等于尿素质量的4%,可起到调节溶液酸性的作用,能够抑制过氧化氢的离解,如若枸橼酸加入量过大,第一,可能会致原料中残留枸橼酸过多,达4%以上的枸橼酸残留能够致小鼠死亡,本方法所制得的产物中的枸橼酸的残留含量为0.05~0.06%,安全性得到很大提升;第二,过氧化碳酰胺在强酸性条件下容易分解,导致其稳定性受到影响。
[0044]
在一个可选的实施方式中,混合包括:
[0045]
将依地酸二钠加入过氧化氢溶液,然后再加入枸橼酸得到第一料液,将尿素加入第一料液中,得到混合体系;
[0046]
在一个可选的实施方式中,依地酸二钠的用量为尿素的质量的 0.1%~1.0%;
[0047]
可选的,依地酸二钠的用量可以为尿素的质量的0.1%、0.2%、0.3%、 0.4%、0.5%、0.6%、0.7%、0.8%、0.9%以及1.0%之间的任意值;
[0048]
在一个可选的实施方式中,过氧化氢溶液与尿素的投料摩尔比为 1:1~1.1:1。
[0049]
可选的,过氧化氢溶液与尿素的投料摩尔比可以为1:1、1.05:1以及1.1:1 之间的任意值。
[0050]
依地酸二钠可以络合金属离子,防止过氧化氢分解,其用量过低可能会致达不到络合金属离子效果,用量过高会致引入太多其他物质,这对于产品的纯度来说是不利的。
[0051]
在一个可选的实施方式中,反应包括:将混合体系在30~40℃条件下保温5~30min,得到第二料液。
[0052]
可选的,混合体系的温度可以为30℃、31℃、32℃、33℃、34℃、35℃、 36℃、37℃、38℃、39℃以及40℃之间的任意值,保温的时间可以为5min、 10min、15min、20min、25min以及30min之间的任意值。
[0053]
混合体系温度不低于30度是为了加速尿素的溶解,若高于40度则会导致过氧化氢的分解。
[0054]
在一个可选的实施方式中,析晶处理包括:将体系先冷却至10℃,然后再冷却至-5℃,维持体系在-7℃至0℃搅拌析晶10~12h;
[0055]
在一个可选的实施方式中,析晶处理之前包括:将体系使用0.45μm聚丙烯滤膜进行过滤。
[0056]
可选的,析晶处理时,体系的温度可以为-7℃、-6℃、-5℃、-4℃、-3℃、
ꢀ‑
2℃、-1℃以及0℃之间的任意值,搅拌析晶的时间可以为10h、10.5h、11h、 11.5h以及12h之间的任意
值。
[0057]
自然结晶后形成的物质整块沉底,不利于后续过滤除母液操作,低温搅拌析晶能够获得细腻的结晶颗粒,能够很好的进行过滤除母液操作,故选择搅拌结晶模式。一次结晶收得的湿品,需等与母液再次过夜重结晶的湿品合并才能进入真空干燥工序,一次结晶湿品的暂存时间偏长,不利于生产效率的提高,且二次结晶可能会引入更多的杂质。因此选择一次结晶模式。析晶温度设置在-7℃~0℃,首先能够提高过氧化碳酰胺原料的收率,其次能够使碳酰胺和过氧化氢结合的更加牢固,提高其稳定性。
[0058]
在一个可选的实施方式中,过滤包括:将体系用200~400目尼龙筛布过滤,得到过氧化碳酰胺湿品。
[0059]
可选的,尼龙筛布的规格可以为200目、250目、300目、350目以及 400目之间的任意值。
[0060]
在一个可选的实施方式中,干燥采用真空干燥,温度为30~40℃,时间为24~36h,真空度大于等于0.09mpa。
[0061]
可选的,真空干燥的温度可以为30℃、31℃、32℃、33℃、34℃、35℃、 36℃、37℃、38℃、39℃以及40℃之间的任意值,时间可以为24h、25h、 26h、27h、28h、29h、30h、31h、32h、33h、34h、35h以及36h之间的任意值。
[0062]
干燥温度高于40℃时,会影响产品的稳定性,温度过低时,又难以干燥,因此对干燥温度和时间的优选,十分重要。
[0063]
本技术还提供一种过氧化碳酰胺,使用本技术提供的过氧化碳酰胺的制备方法制得。
[0064]
本技术提供的制备方法所制得的过氧化碳酰胺,其安全性和稳定性更好,有利于其保存和后续的安全使用。
[0065]
本技术还提供一种注射用过氧化碳酰胺制剂的制备方法,包括:
[0066]
将本技术提供的过氧化碳酰胺用注射用水溶解,然后过滤除菌、冻干得到注射用过氧化碳酰胺制剂。
[0067]
在一个可选的实施方式中,注射用水的温度为30~40℃;
[0068]
在一个可选的实施方式中,过滤除菌包括:
[0069]
依次用0.45μm聚醚砜筒式过滤器减菌过滤、用0.22μm聚醚砜筒式过滤器进行两次除菌过滤;
[0070]
在一个可选的实施方式中,第一次用0.22μm聚醚砜筒式过滤器进行除菌过滤在c级环境局部a级条件下进行;第二次用0.22μm聚醚砜筒式过滤器进行除菌过滤在b级环境局部a级条件下进行;
[0071]
在一个可选的实施方式中,冻干包括:在-40~-50℃条件下预冻干1~5h,然后在3h内升温至1℃,保温6~8h;再升温至15℃,保温2~4h;再升温至22℃,保温2~4h。
[0072]
可选的,注射用水的温度可以为30℃、31℃、32℃、33℃、34℃、35℃、 36℃、37℃、38℃、39℃以及40℃之间的任意值;
[0073]
可选的,预冻干的温度可以为-40℃、-41℃、-42℃、-43℃、-44℃、-45℃、
ꢀ‑
46℃、-47℃、-48℃、-49℃以及-50℃之间的任意值,预冻干的时间可以为 1h、2h、3h、4h以及5h之间的任意值,保温的时间可以为6h、7h以及8h 之间的任意值,再升温之后保温的时间可以
为2h、3h以及4h之间的任意值。
[0074]
使用冻干工艺制备注射用过氧化碳酰胺制剂,产品稳定性提高,不再出现炸瓶现象,产品安全性得到提升,同时采用多重除菌过滤工艺,去除了体系中的多种杂质,所得产物纯度更高,稳定性更好。
[0075]
本技术还提供一种注射用过氧化碳酰胺制剂,使用本技术提供的注射用过氧化碳酰胺制剂的制备方法制得。
[0076]
本技术提供的制备方法所制得的注射用过氧化碳酰胺制剂,其杂质少,稳定性好,安全性高。
[0077]
实施例1
[0078]
s1:将含17kg过氧化氢的溶液转移至100l溶解罐中,开启搅拌,投入66g依地酸二钠,搅拌溶解。然后加入412g枸橼酸,搅拌溶解,即得料液1;
[0079]
s2:开启溶解罐水浴加热,将30kg尿素投入料液1中,控制水浴温度30℃,搅拌溶解后,继续30℃水浴保温5分钟,即得料液2;
[0080]
s3:料液2经0.45μm聚丙烯滤膜过滤至c级洁净区的100l结晶罐中。开启结晶罐搅拌,打开夹套冷冻水系统相应阀门降温析晶。待析晶料液冷却至10℃,排去夹套冷冻水,开启低温循环泵,夹套转入液继续降。待析晶料冷却至-5℃开始析晶计时,维持析晶料液-7℃搅拌析晶 10h。析晶结束后,将析晶料液转入过滤器中,经200目尼龙筛布过滤,至基本无母液滤出时得过氧化碳酰胺湿品;
[0081]
s4:将过氧化碳酰胺湿品装盘(湿品装盘厚度应≤2cm),按从上至下顺序送入真空干燥机,开启水箱加热和真空泵,控制真空度≥ 0.09mpa,控制加热用水温度,控制加热用水温度,控制加热用水温度 30℃,控制真空干燥时间24h(加热用水温度升至30℃开始计时),即得过氧化碳酰胺干品;
[0082]
s5:在配制罐中加入理论量50%注射用水(30℃),加入已称量好的过氧化碳酰胺,搅拌使其完全溶解,补加注射用水定容至全量;
[0083]
s6:配制好的药液用0.45μm聚醚砜筒式过滤器减菌过滤后,在c级环境局部a级条件下用已经灭菌的0.22μm聚醚砜筒式过滤器过滤除菌,然后在b级环境局部a级条件下用已经灭菌的0.22μm聚醚砜筒式过滤器进行除菌过滤灌装于玻璃瓶中;
[0084]
s7:冻干:预冻:-45℃条件下维持3h;第一段:3h内缓慢升温至1 度,保温7h;第二段:升温至15℃,保温3h;第三段:升温至22℃,保温3h;
[0085]
s8:压塞,轧盖,得到注射用过氧化碳酰胺制剂。
[0086]
实施例2
[0087]
s1:将含17kg过氧化氢的溶液转移至100l溶解罐中,开启搅拌,投入30g依地酸二钠,搅拌溶解。然后加入1.2kg枸橼酸,搅拌溶解,即得料液1;
[0088]
s2:开启溶解罐水浴加热,将30kg尿素投入料液1中,控制水浴温度35℃,搅拌溶解后,继续35℃水浴保温20分钟,即得料液2;
[0089]
s3:料液2经0.45μm聚丙烯滤膜过滤至c级洁净区的100l结晶罐中。开启结晶罐搅拌,打开夹套冷冻水系统相应阀门降温析晶。待析晶料液冷却至10℃,排去夹套冷冻水,开启低温循环泵,夹套转入液继续降。待析晶料冷却至-5℃开始析晶计时,维持析晶料液-4℃搅拌析晶 11h。析晶结束后,将析晶料液转入过滤器中,经300目尼龙筛布过滤,至基本无母
液滤出时得过氧化碳酰胺湿品;
[0090]
s4:将过氧化碳酰胺湿品装盘(湿品装盘厚度应≤2cm),按从上至下顺序送入真空干燥机,开启水箱加热和真空泵,控制真空度≥ 0.09mpa,控制加热用水温度,控制加热用水温度,控制加热用水温度 35℃,控制真空干燥时间30h(加热用水温度升至35℃开始计时),即得过氧化碳酰胺干品;
[0091]
s5:在配制罐中加入理论量50%注射用水(35℃),加入已称量好的过氧化碳酰胺,搅拌使其完全溶解,补加注射用水定容至全量;
[0092]
s6:同实施例1;
[0093]
s7:冻干:预冻:-50℃条件下维持1h;第一段:3h内缓慢升温至1 度,保温6h;第二段:升温至15℃,保温2h;第三段:升温至22℃,保温2h;
[0094]
s8:压塞,轧盖,得到注射用过氧化碳酰胺制剂。
[0095]
实施例3
[0096]
s1:将含17kg过氧化氢的溶液转移至100l溶解罐中,开启搅拌,投入300g依地酸二钠,搅拌溶解。然后加入303g枸橼酸,搅拌溶解,即得料液1;
[0097]
s2:开启溶解罐水浴加热,将30kg尿素投入料液1中,控制水浴温度40℃,搅拌溶解后,继续40℃水浴保温30分钟,即得料液2;
[0098]
s3:料液2经0.45μm聚丙烯滤膜过滤至c级洁净区的100l结晶罐中。开启结晶罐搅拌,打开夹套冷冻水系统相应阀门降温析晶。待析晶料液冷却至10℃,排去夹套冷冻水,开启低温循环泵,夹套转入液继续降。待析晶料冷却至-5℃开始析晶计时,维持析晶料液0℃搅拌析晶 12h。析晶结束后,将析晶料液转入过滤器中,经400目尼龙筛布过滤,至基本无母液滤出时得过氧化碳酰胺湿品;
[0099]
s4:将过氧化碳酰胺湿品装盘(湿品装盘厚度应≤2cm),按从上至下顺序送入真空干燥机,开启水箱加热和真空泵,控制真空度≥ 0.09mpa,控制加热用水温度,控制加热用水温度,控制加热用水温度 40℃,控制真空干燥时间36h(加热用水温度升至40℃开始计时),即得过氧化碳酰胺干品;
[0100]
s5:在配制罐中加入理论量50%注射用水(40℃),加入已称量好的过氧化碳酰胺,搅拌使其完全溶解,补加注射用水定容至全量;
[0101]
s6:同实施例1;
[0102]
s7:冻干:预冻:-40℃条件下维持5h;第一段:3h内缓慢升温至1 度,保温8h;第二段:升温至15℃,保温4h;第三段:升温至22℃,保温4h;
[0103]
s8:压塞,轧盖,得到注射用过氧化碳酰胺制剂。
[0104]
实施例4
[0105]
与实施例1不同的是,s1中投入依地酸二钠的量为150g,加入枸橼酸的量为498g。
[0106]
实施例5
[0107]
与实施例1不同的是,s1中使用的过氧化氢的质量为18.7kg。
[0108]
对照例1
[0109]
分别取枸橼酸含量为1%,2%,4%,6%,8%的注射用过氧化碳酰胺对小鼠进行急性毒性试验。
[0110]
试验给药方式为实验兔耳缘静脉滴注。选用检疫合格的km小鼠50只,雌雄各半,分
为5组,每组10只,按含1%枸橼酸,2%枸橼酸,4%枸橼酸, 6%枸橼酸,8%枸橼酸的注射用过氧化碳酰胺分别编为d1,d2,d3,d4, d5组,作为供试品组。每只小鼠尾静脉注射上述含不同浓度枸橼酸的注射用过氧化碳酰胺(5mg/ml),给药体积为0.5ml/只,1次/天,共给1次,推注速度均为0.05ml/s。每天肉眼观察小鼠的状态,是否死亡。给药后72h 确认小鼠是否死亡。
[0111]
试验结果:试验期间d1按给药体积为0.5ml/只,推注速度为0.05ml/s,给药后动物的外观体征、行为活动和一般状况未出现异常,72h后也无异常; d2按给药体积为为0.5ml/只,推注速度为0.05ml/s,给药后动物的外观体征、行为活动和一般状况未出现异常,72h后也无异常;d3按给药体积为为0.5ml/只,推注速度为0.05ml/s,给药后其中4只小鼠抽搐,精神状态不好,72h后4只死亡,其它正常。d4按给药体积为为0.5ml/只,推注速度为0.05ml/s,给药后其中4只小鼠抽搐死亡,72h后6只死亡;d5按给药体积为为0.5ml/只,推注速度为0.05ml/s,给药后其中10只小鼠全部抽搐死亡。
[0112]
由对照例1可知,含4%枸橼酸以上浓度的注射用过氧化碳酰胺会致小鼠死亡,为保证产品中枸橼酸的残留量低于4%,制备工艺中枸橼酸的添加量应不超过4%,以提高产品的安全性。
[0113]
对照例2
[0114]
反应体系中不同添加量的枸橼酸对产物收率的影响如下表1所示:
[0115]
表1不同枸橼酸添加量所得产品收率数据
[0116][0117]
由对照例2可知,枸橼酸添加量较现有技术降低后,并不影响产品收率,枸橼酸的添加量并不是越多越好,加入过量的酸反而引起过氧化氢分解,并且,结合对照例1可知,控制枸橼酸的加入量便于减少最终在原料中的枸橼酸的残留量,更利于提高产品的安全性。
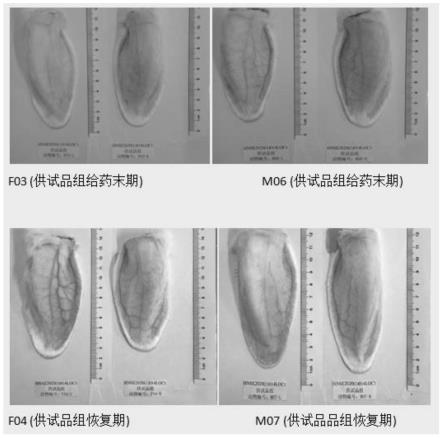
[0118]
对照例3
[0119]
在s3进行搅拌冷却析晶时,不同的析晶温度对产品的收率以及产品质量的影响,结果如下表2所示:
[0120]
表2不同析晶温度所得产品收率数据
[0121]
析晶温度析晶状态收率ph含量0~4℃细腻结晶颗粒56.8%3.834.2%-4℃~0℃细腻结晶颗粒77.2%3.635.1%-8℃~-4℃细腻结晶颗粒78.8%3.634.8%-12℃~-8℃液面出现结冰状况,影响搅拌———
[0122]
由对照例3可知,当析晶温度为0~4℃时,收率较低,-12℃~-8℃液面出现结冰状况,不能进行搅拌,0-4℃和-4℃-~-8℃两个时间段能够获得较好产品收率和质量,因此本
申请提供的0℃~-7℃作为析晶温度时所得产品收率高,质量好。
[0123]
对照例4
[0124]
s4真空干燥的温度不同,当铺盘厚度为2cm时,产品性能的变化如下表3所示:
[0125]
表3不同干燥温度产品的状态
[0126]
温度干燥24h的干燥失重干燥36h的干燥失重ph含量30℃0.9%0.5%3.635.1%35℃0.6%0.3%3.635.4%40℃0.5%0.2%3.635.3%45℃0.2%0.2%3.932.8%
[0127]
由对照例4可知,当真空干燥的温度处于30-40℃时,产品中过氧化氢的含量相对温度且保持在较高水平,但当温度达到45℃时,其中过氧化氢的含量显著下降,产品出现不稳定状态,因此,本技术提供的干燥温度效果更优。
[0128]
对实施例所得产品中有效物质及其含量进行检测,具体方法及结果如下,其中样品来自本实施例所制得的产品:
[0129]
(1)仪器与设备
[0130]
天平:梅特勒-托利多ma105du,高效液相色谱仪:岛津lc-20a
[0131]
表4对照品信息
[0132]
名称批号含量来源浓过氧化氢溶液b210417131.0%西陇科学股份有限公司
[0133]
色谱条件与系统适应性
[0134]
照高效液相色谱法(通则0512)试验。用十八烷基硅烷键合硅胶为填充剂,以45%乙腈为流动相,流速为每分钟0.5ml,检测波长为191nm,柱温为30℃。取对照品溶液20μl注入液相色谱仪,记录色谱图,理论板数按过氧化氢峰计算不低于3000。
[0135]
流动相:45%乙腈。
[0136]
对照品溶液:精密称取浓过氧化氢溶液1)0.2342g,2)0.2423g置100ml 量瓶中,用45%乙腈稀释至刻度,摇匀,在精密量取1.0ml置10ml量瓶中,加45%乙腈稀释至刻度,混匀。以过氧化氢计,浓度分比为1)0.07260mgml 2)0.07511mg/ml。
[0137]
测定法:精密量取对照品溶液与供试品溶液各20μl,分别注入液相色谱仪,记录色谱图。
[0138]
计算公式:过氧化氢(%)=供试品溶液峰面积
÷
对照品溶液峰面积
×
对照品溶液浓度
×
稀释倍数
÷
取样量
×
100%
[0139]
限度:按外标法以峰面积计算,按干燥品计,含过氧化氢(h2o2)应为30.0%~36.0%。
[0140]
(2)专属性试验
[0141]
1)空白溶液:即流动相。
[0142]
2)过氧化碳酰胺溶液:精密称定过氧化碳酰胺0.2540g置100ml量瓶中,用45%乙腈稀释至刻度,摇匀,在精密量取1.0ml置10ml量瓶中,加 45%乙腈稀释至刻度,混匀。
[0143]
3)对照品溶液:精密称取浓过氧化氢溶液0.2487g置100ml量瓶中,用45%乙腈稀释至刻度,摇匀,在精密量取1.0ml置10ml量瓶中,加45%乙腈稀释至刻度,混匀。
[0144]
进样:分别取空白溶液、过氧化碳酰胺溶液、对照品溶液各进一针,记录色谱图。
[0145]
结果:空白溶剂(稀释剂)应对含量检测无干扰,过氧化氢峰理论塔板数应不小于3000,过氧化氢峰拖尾应小于1.5;尿素峰与过氧化氢峰分离度应大于等于1.5。
[0146]
结论:专属性符合要求。
[0147]
表5专属性试验结果
[0148][0149][0150]
(3)灵敏度试验
[0151]
溶液配制
[0152]
1)定量限溶液稀释过程:取对照品溶液1.0ml,加流动相稀释至200ml,再取1ml稀释至2ml。
[0153]
2)检测限溶过程:取定量限溶液1ml,加流动相稀释至3ml。
[0154]
试验结果
[0155]
定量限溶液:当对照品溶液浓度为1.815μl/ml,对照品峰信噪比为11.01;
[0156]
检测限溶液:当对照品溶液浓度为0.605μl/ml,对照品峰信噪比为4.55。
[0157]
结论:符合要求。
[0158]
(4)精密度试验
[0159]
重复性
[0160]
供试品溶液:取供试品1)0.2613g,2)0.2527g,3)0.2507g,4)0.2635g, 5)0.2553g,6)0.2563g,精密称定,置100ml量瓶中,用45%乙腈稀释至刻度,摇匀,在精密量取1.0ml置10ml量瓶中,加45%乙腈稀释至刻度,混匀。
[0161]
对照品溶液:精密称取浓过氧化氢溶液1)0.2342g,2)0.2423g置100ml 量瓶中,用45%乙腈稀释至刻度,摇匀,在精密量取1.0ml置10ml量瓶中,加45%乙腈稀释至刻度,混匀。以过氧化氢计,浓度分比为1)0.07260mgml 2)0.07511mg/ml。
[0162]
进样:用空白溶液和对照品溶液建立系统适应性,合格后,各重复性溶液分别进样一次。
[0163]
结果:计算6份重复性溶液的含量,其结果rsd应≤2.0%。
[0164]
结论:重复性良好,符合要求。
[0165]
表6重复性试验结果
[0166]
序号123456rsd(%)含量34.7%34.8%36.0%35.4%36.1%35.5%1.5%
[0167]
中间精密度试验
[0168]
由另一名检验员操作,取供试品1)0.2519g,2)0.2501g,3)0.2565g,4)0.2513g,5)0.2635g,6)0.2582g,精密称定,置100ml量瓶中,用45%乙腈稀释至刻度,摇匀,在精密量取
1.0ml置10ml量瓶中,加45%乙腈稀释至刻度,混匀。
[0169]
对照品溶液:精密称取浓过氧化氢溶液1)0.2342g,2)0.2423g置100ml 量瓶中,用45%乙腈稀释至刻度,摇匀,在精密量取1.0ml置10ml量瓶中,加45%乙腈稀释至刻度,混匀。以过氧化氢计,浓度分比为1)0.07260mgml 2)0.07511mg/ml。
[0170]
进样:用空白溶液和对照品溶液建立系统适应性,合格后,各重复性溶液分别进样一次。
[0171]
结果:计算6份重复性溶液的含量,其结果rsd应≤2.0%,计算12 份重复性溶液的含量,其结果rsd应≤2.0%。
[0172]
结论:符合要求。
[0173]
表7中间精密度试验结果
[0174]
序号1234566针rsd(%)含量35.5%35.7%35.8%35.0%35.2%35.1%0.92%序号78910111212针rsd(%)含量34.7%34.8%36.0%35.4%36.1%35.5%1.2%
[0175]
(5)线性范围
[0176]
1)对照品贮备液:精密称取过氧化氢溶液(经标定含量)0.2487g,置100ml量瓶中,用45%乙腈稀释至刻度,摇匀,作为对照品贮备液。
[0177]
2)线性系列溶液:
[0178]
表8线性系列溶液配置浓度
[0179][0180]
进样:精密量取上述线性系列溶液各20μl,分别注入液相色谱仪,记录色谱图。
[0181]
结果:以浓度为横坐标,峰面积为纵坐标,制作标准曲线,进行一次线性回归,相关系数应≥0.995。
[0182]
结论:符合要求。
[0183]
表9线性试验结果
[0184][0185]
(6)溶液稳定性
[0186]
取供试品溶液进行试验:
[0187]
进样:分别在供试品溶液放置的0、2、4、6、8小时后进行检测,记录色谱图。
[0188]
结果:供试品溶液中过氧化氢峰面积rsd应≤3.0%。
[0189]
结论:符合要求。
[0190]
表10溶液稳定性试验结果
[0191]
时间(h)02468rsd峰面积136295313219351283285125901612604883.42%
[0192]
(7)准确度试验
[0193]
溶液制备
[0194]
加标重复性溶液:取供试品1)0.2502g,2)0.2803g,3)0.2701g,4) 0.2578g,5)0.2705g,6)0.2614g,精密称定,置100ml量瓶中,用45%乙腈稀释至刻度,摇匀,精密量取1.0ml置10ml量瓶中,再精密加入对照品贮备液(精密称取过氧化氢溶液(经标定含量)0.2487g,置100ml量瓶中,用45%乙腈稀释至刻度,摇匀,作为对照品贮备液。)1.0ml,加45%乙腈稀释至刻度,混匀。
[0195]
对照品溶液:同上。
[0196]
进样:分别精密量取上述6份溶液各20μl注入高效液相色谱仪,记录色谱图。
[0197]
结果:计算回收率,回收率应在92%~102%,rsd应≤2%。
[0198]
回收率(%)=(测得量-本地量)
÷
过氧化氢加入量
×
100%。
[0199]
过氧化碳酰胺中过氧化氢含量测定
[0200]
表11过氧化氢回收率结果
[0201]
[0202][0203]
该过氧化碳酰胺中过氧化氢含量的测定方法,对每个参数设立可接受标准和验证步骤,通过验证证明该检验方法的系统适用性、专属性、准确性、精密度(包括重复性和重现性)、线性、检测限、定量限、溶液稳定性和耐用性以确认本方法的可靠性和准确性,适合过氧化碳酰胺中过氧化氢含量的检测。
[0204]
使用该测定方法测得的本实施例产品中过氧化氢的含量,即药物的有效成分含量较高,稳定性很好。
[0205]
为了考察本技术提供的注射用过氧化碳酰胺的质量,发明人对实施例所得产品的性能做了如下试验。
[0206]
一、安全性试验
[0207]
按照国家药品监督管理局颁布的《药物非临床研究质量管理规范》(2017)、《药物刺激性、过敏性和溶血性研究技术指导原则》(2014)和《非临床安全性评价供试品检测要求的q&a》,thenationalacademics(washingtond.c.)颁布的《guideforthecareanduseoflaboratoryanimals(eighthedition)》(2011)的规定,进行多次给药对实验兔的血管刺激性反应、对实验兔体外红细胞的溶血性反应、对豚鼠主动全身过敏性反应以及对大鼠的被动皮肤过敏反应进行测试,结果均符合要求,没有异常反应,安全性可靠。具体试验及数据结果如下:
[0208]
1)多次给药实验兔的血管刺激性反应:试验给药方式为实验兔耳缘静脉滴注。选用检疫合格的实验兔8只,雌雄各半,作为供试品组(动物编号为雌性f01~f04、雄性m05~m08)。每只实验兔左侧耳缘静脉滴注注射用过氧化碳酰胺(10.0mg/ml),右侧耳缘静脉滴注同体积的5%葡萄糖注射液作为对照。给药体积为2.0ml/kg,1次/天,共给5天,推注速度均为1.7ml/min。每天肉眼观察注射部位的血管及周围组织有无肿胀、充血、渗出、变性或坏死等的变化。末次给药后72h取4只实验兔后肢隐静脉注射丙泊酚乳状注射液(15mg/kg)麻醉后腹主动脉放血安乐死,进行组织病理学检查。4只实验兔继续观察14天,同法肉眼观察注射部位的血管及周围组织的变化,并进行组织病理学检查,以了解刺激性反应的可逆程度。
[0209]
试验结果:试验期间动物按给药体积为2.0ml/kg,推注速度为1.7ml/min,每天1次,连续5天,给药后动物的外观体征、行为活动和一般状况均未出现异常,注射部位血管及周围组织无肿胀、充血、渗出、变性或坏死等变化,且未出现死亡。大体观察和组织病理学检查结果显示注射用过氧化碳酰胺给药侧与阴性对照侧耳缘静脉均无血管刺激性反应,如图1所示。
[0210]
结论:在本试验条件下,注射用过氧化碳酰胺对实验兔未见明显的血管刺激性反应。
[0211]
2)实验兔体外红细胞的溶血性反应:采用了注射用过氧化碳酰胺的常用静脉滴注浓度(10.0mg/ml)进行试验。取试管7支,进行编号,1~5号管为供试品管,6号管为阴性对照管,7号管为阳性对照管。依次加入2%红细胞悬液和5%葡萄糖注射液、纯水,混匀后,立即置37℃水浴箱放置 30min,然后分别加入不同量的药液。摇匀后,置37℃水浴箱中观察3h, 1h内每隔15min观察一次。2~3h,每隔1h观察一次反应结果,3h后将各管的溶液置入干燥离心管中离心,测试各管吸光光度值。
[0212]
试验结果:阴性对照5%葡萄糖注射液管红细胞下沉,上层液体无色澄明,结果为无溶血;观察结束时振摇管底沉淀,红细胞可分散,表明无红细胞凝聚。阳性对照纯水管溶液澄明红色,管底无红细胞残留,结果为完全溶血。注射用过氧化碳酰胺(10.0mg/ml)各管均红细胞下沉,上层液体无色澄明,结果为无溶血;观察结束时振摇管底沉淀,红细胞可分散,表明无红细胞凝聚。分光光度法显示注射用过氧化碳酰胺供试品(10.0mg/ml) 第1~5管溶血率均小于5%,如图2所示。
[0213]
结论:在本试验条件下,注射用过氧化碳酰胺(批号:191115-2) (10.0mg/ml)对实验兔体外红细胞未见明显的溶血性和红细胞凝聚反应。
[0214]
3)豚鼠主动全身过敏反应:选择检疫合格的24只豚鼠,雌雄各半,体重300.5~389.0g,按性别、体重采用随机数字方法分为4组,分别为阴性对照组(5%葡萄糖注射液)、阳性对照组(100mg/ml卵蛋白液)、供试品低剂量组(5.0mg/ml)及供试品高剂量组(10.0mg/ml),每组6只动物。各组腹腔注射给予相应的药物,隔天一次,共3次。致敏给药体积为0.5ml/ 只。致敏末次给药后第14天和第21天分别经足趾静脉注射给予与致敏时等浓度药液进行攻击,攻击剂量为致敏剂量的2倍,攻击体积1.0ml/只。激发后立即观察30min,详细观察每只动物的反应,症状的出现及消失时间。观察3h,根据过敏反应发生率和发生程度进行综合判断。
[0215]
试验结果:阴性对照组过敏反应为阴性,阳性对照组过敏反应呈阳性,注射用过氧化碳酰胺低剂量组和高剂量组豚鼠静脉激发后3h内全部动物均未出现过敏性反应症状,过敏反应为阴性。
[0216]
结论:在本试验条件下,注射用过氧化碳酰胺(批号:191115-2)未引起豚鼠的全身过敏反应。
[0217]
4)大鼠被动皮肤过敏反应:试验分两部分:致敏及抗体制备和皮肤被动致敏及激发。致敏阶段设阴性对照组(5%葡萄糖注射液)和阳性对照组 (100mg/ml卵蛋白液)、供试品低剂量组(10.0mg/ml)及供试品高剂量组 (20.0mg/ml),每组2只动物。各组腹腔注射相应药液,0.5ml/只,隔天给药一次,共5次。末次致敏给药后第13天经腹主动脉采血,分离血清,
ꢀ‑
20℃储存备用。
[0218]
皮肤被动致敏及激发也分4组,分别与致敏阶段分组相对应,每组雌雄各3只动物。各组在预先剪毛的左侧及右侧背部皮肤的下部、中部、上部位置分别皮内注射相应组别抗血清原液,1:2抗血清和1:4抗血清各0.1ml,两点间距离1.0cm,进行被动致敏。
[0219]
被动致敏24h后,各组静脉注射与致敏剂量相同的激发抗原加等体积的1.0%伊文斯蓝染料进行激发。激发后30min将动物麻醉后颈椎脱臼处死,剪取背部皮肤,用游标卡尺测量皮肤内层的斑点直径大小;不规则斑点的直径为长径与短径之和的一半。斑点直径大于5mm者判断为阳性。
[0220]
试验结果:阴性对照组5/6出现蓝斑,但斑点的直径均小于5mm,呈阴性;阳性对照组4/6出现蓝斑,斑点的直径均大于5mm,呈阳性;供试品低剂量组1/6出现蓝斑,但斑点的直径小于5mm,呈阴性;供试品高剂量组3/6出现蓝斑,但斑点的直径小于5mm,呈阴性,如图3所示。
[0221]
结论:在本试验条件下,注射用过氧化碳酰胺供试品(批号:191115-2) 未引起大鼠的被动皮肤过敏反应。
[0222]
二、稳定性试验
[0223]
本技术提供的过氧化碳酰胺原料和注射用过氧化碳酰胺稳定性好,通过稳定性试验评价其结果如下:
[0224]
取三批过氧化碳酰胺原料在2~8度条件下经过24个月长期稳定性试验考察,各项指标均稳定,结果均符合要求。且枸橼酸的残留量为0.05%~0.06%范围内,依地酸二钠的残留量范围为0.0001%以下。
[0225]
表12过氧化碳酰胺稳定性第一批次试验结果
[0226][0227][0228]
表13过氧化碳酰胺稳定性第二批次试验结果
[0229][0230]
表14过氧化碳酰胺稳定性第三批次试验结果
[0231][0232]
取三批注射用过氧化碳酰胺制剂在4~8度经过24个月长期稳定性实验考察,各项指标均稳定,结果均符合要求。
[0233]
表15过氧化碳酰胺制剂稳定性第一批次试验结果
[0234][0235][0236]
表16过氧化碳酰胺制剂稳定性第二批次试验结果
[0237][0238]
表17过氧化碳酰胺制剂稳定性第三批次试验结果
[0239]
[0240][0241]
通过实施例产品的试验和检测结果可知,本技术提供的制备过氧化碳酰胺的方法,以及使用其制备所得的过氧化碳酰胺、注射用过氧化碳酰胺制剂的安全性更好,稳定性更高。
[0242]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1