一种盐酸多巴酚丁胺的制备方法与流程
1.本发明涉及一种盐酸多巴酚丁胺的制备方法领域,特别是将盐酸多巴酚丁胺粗品特定方法纯化干燥。
背景技术:
2.盐酸多巴酚丁胺(dobutamine hydrochloride)为多巴胺同系物,为一选择性心脏β1-受体兴奋剂。盐酸多巴酚丁胺正性肌力作用比多巴胺强,对β2-受体和α-受体兴奋性较弱。治疗量能增加心肌收缩力,增加心排血量,很少增加心肌耗氧量,可降低外周血管阻力,能降低心室充盈压,促进房室结传导。
3.目前盐酸多巴酚丁胺合成主要是以3,4-二甲氧基苯乙胺和4-(4-甲氧苯基)-2-丁酮为起始原料,经过缩合、加氢、成盐反应后得到中间体n-(3,4-二甲氧基苯乙基)-4-(4-甲氧苯基)丁-2-胺盐酸盐(化合物v),化合物v经过脱甲基并精制后得到盐酸多巴酚丁胺。其中化合物v为盐酸多巴酚丁胺最重要的中间体之一,经过脱甲基并盐酸成盐后即可以得到盐酸多巴酚丁胺。上海第二制药厂以3,4-二甲氧基苯乙胺和4-(4-甲氧苯基)-2-丁酮为原料,对甲苯磺酸催化下,苯回流分水,脱水缩合得到化合物iii,采用硼氢化钾还原,乙醚萃取,乙醚盐酸溶液成盐得到化合物v。目前的工艺中合成过程会引入多种毒性的苯系物(如对甲基苯磺酸等),生产中需要严格的控制限度,也增加了工艺除杂的难度。
4.盐酸多巴酚丁胺的析晶条件较为苛刻,通常以随机方式结晶,常常出现油状物,也导致无法观察到的杂质结晶形成。cn104860833a公开了盐酸多巴酚丁胺的纯化方法,包括粗品与甲醇混合加热搅拌加入溶剂a(水、乙醇或丙酮),冷却析晶真空干燥,该工艺需要真空干燥,才能将甲醇/丙酮等溶剂清除,在实际生产中收率不高,并且获得的晶体虽然纯度高,但其晶体颗粒大,没有规则的形状,有变色等问题,需要进一步加工才能作为药物制剂原料,否则会给临床用药安全带来挑战。日本公司专利us5073648a公开了一种盐酸多巴酚丁胺的纯化方法,将粗品放入一定盐酸中,加热溶解后室温条件自然冷却过夜,析晶后烘干,得到纯度98.3%收率84%的产品,该工艺获得产品的纯度和收率有限,在制剂中还需要进一步纯化,并且同样存在上述问题。盐酸多巴酚丁胺在注射剂配置过程中需要避光处理,较为严重的情况下必须加入抗氧化剂或即配即用。
技术实现要素:
5.针对现有技术存在的问题,本发明在对盐酸多巴胺纯化的粗品过程中发现采取特殊溶剂介质,通过通入氮气并挥发溶剂,而不是旋转蒸发或单纯的溶剂沉淀法,在一定的条件下可以制备成制剂用的原研药。制备条件包括但不限于控制温度和转速的变化、氮气通入速率的变化,在溶剂挥发浓缩后补入适量水分等等;例如将搅拌过程转速控制在15-75rpm,温度30-65℃,并随时间和溶剂变化加热或冷却,通过网带快速干燥,在一定时间条件下,可获得粒径分布均匀的具有规则形状的光滑晶体,且硬度较好不易结块,特别是光稳定性提高,适合药物生产和临床配置。
6.本发明人在试验中发现导致析晶不稳定的部分原因是溶剂和纯化工艺问题,例如制备过程中带入的苯系物杂质,晶体的结构等,如果采用甲醇作为溶剂会产生一部分混合晶型现象;而以现有技术的真空干燥技术,晶体粒径分布方差较大,吸湿性也不理想。另外干燥条件、温度和搅拌速度也是结晶过程中的必要因素,本发明的产品便于制剂生产,并发现其变色时间有所延迟,这对临床用药带来较大便利。
7.本发明技术方案如下:
8.步骤1:将盐酸多巴酚丁胺粗品加入乙醇中,搅拌后再加入石油醚,缓慢加热并持续搅拌;
9.步骤2:向瓶内通入氮气,优选瓶口以滤纸盖封,溶液搅拌状态下加热至30-60℃;溶液变浑浊后增加搅拌速度,停止加热并自然降温至室温;
10.步骤3:母液减少至原体积的30%-40%时停止搅拌,除去瓶口滤纸,加入水补充母液,至原体积的50%-60%,搅拌至溶液分层后加热至35℃-65℃,并持续通入氮气,直至溶剂至原体积30%-40%时,缓慢冷却至10℃以内,停止搅拌,静置析晶12-24h;
11.步骤4:固液分离,干燥,即得。
12.作为本发明优选方案,步骤1中的多巴酚丁胺粗品和乙醇的质量体积比为1:1.5-3(g/ml),优选为1:2(g/ml);步骤1中的石油醚和乙醇的体积比为1:5-15,优选1:10;
13.作为本发明优选技术方案,步骤1和步骤2中加热速度为1-3℃/min,优选2℃/min;
14.作为本发明优选方案,步骤3中缓慢冷却是以恒定速度冷却至10℃以下,优选地冷却至2-9℃,更优选地为5℃;冷却速度每分钟降低0.5-2℃,优选地为每分钟降低1℃
15.优选地,步骤2中在溶液变浑浊后提高氮气通入速度;优选的从2-3l/min提高到4-5l/min;
16.作为本发明的优选方案,所述石油醚为沸程在30-60℃的石油醚。
17.作为本发明的优选方案,氮气的通入速度为2-5l/min,优选3-4l/min;
18.作为本发明优选方案,所述干燥为网带式烘干,优选三层网带烘干机,更优选热风加热。
19.作为本发明优选方案,热风温度为60-100℃,更优选70-90℃;处理时间1-100min,优选10-60min。
20.作为本发明优选方案,步骤1搅拌速度15-50rpm,优选地为25-40rpm;步骤2中溶液在15-50rpm搅拌速度下加热,溶液浑浊后增加搅拌速度至50-75rpm,优选为60-70rpm;步骤3中在搅拌速度为50-75rpm条件下加热,缓慢冷却是降温速度约1℃/min;
21.更进一步的,步骤1-3在通风橱内操作,或者在具有空气交换设备或通风良好的空间内操作。
22.本发明固液分离方法包括但不限于过滤,离心;
23.本发明乙醇可以为无水乙醇,也可以为95%的乙醇;除非说明书中特殊说明,本发明中的g为重量单位“克,”h为时间单位“小时,”min为时间单位“分钟,”ml为体积单位“毫升,”l为体积单位“升,”rpm为“转/分钟,”室温为22-25℃,常温为20℃。
附图说明
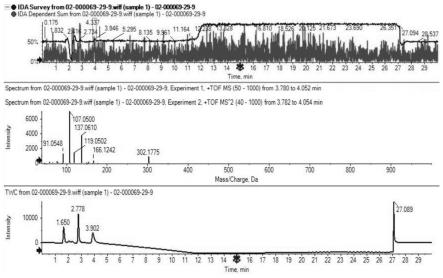
24.图1:实施例1的盐酸多巴酚丁胺检测图谱;
25.图2:实施例1晶体电镜扫描图。
具体实施方式
26.实施例1
27.将100g盐酸多巴酚丁胺粗品加入盛有200ml无水乙醇的反应瓶中,置于通风橱内,以速度30rpm进行磁力搅拌,加入20ml石油醚,以2℃/min的速度进行加热并持续搅拌,向瓶内通入氮气,通入速度为3l/min,瓶口以滤纸盖封,同时加热至35-55℃,溶液逐渐澄清后出现浑浊时,停止加热,增加搅拌速度至60rpm,氮气通入速度增加至4l/min,自然降温至室温(24℃);保持室温,搅拌速度和氮气通入速率,约2-4h后目测母液减少至原体积的1/3左右(约70-75ml)时,停止搅拌,除去瓶口滤纸,补入水,至溶剂为原体积50-55%(110-120ml左右),继续以60rpm速度搅拌,以4l/min速率通入氮气,以2℃/min速度加热,加热至60℃,当溶剂逐渐浓缩至原母液体积35-40%时,停止加热,以1℃/min的速度降温至5℃后停止搅拌,停止通入氮气,静置析晶过夜(18h);固液过滤分离,将沉淀分散置于三层网带烘干桶上,以80℃热风加热10min后即得。
28.检测纯度99.9%(hplc),单杂含量<0.01%,参见检测附图1。单晶结构,粒径≤10μm。
29.实施例2
30.将100g盐酸多巴酚丁胺粗品加入盛有250ml无水乙醇的反应瓶中,置于装有排风系统的室内,以速度35rpm进行磁力搅拌,加入25ml石油醚,以2℃/min的速度进行加热至35-55℃,持续搅拌直至溶液逐渐澄清后出现浑浊且有气体挥发时,停止加热,逐渐以1℃/min左右的速率来降低温度直至室温;保持室温,和搅拌速度,以3l/min速度通入氮气,约2-3h后目测母液减少至原体积的1/3左右(约90-95ml)时,停止搅拌,补入水,至溶剂为原体积50-55%(130-140ml左右),继续以35rpm速度搅拌,以2℃/min速度加热,加热至60℃,当溶剂逐渐浓缩至原母液体积40%左右时,停止加热,以1℃/min的速度降温至5℃后停止搅拌,密封瓶口,静置析晶过夜(18h);固液过滤分离,将沉淀分散置于三层网带烘干桶上,以90℃热风加热10min后即得。
31.检测纯度99.6%(hplc),单杂含量<0.02%,检测图谱和实施例1相同。单晶结构。
32.实施例3
33.将100g盐酸多巴酚丁胺粗品加入盛有200ml无水乙醇的反应瓶中,置于通风良好的空间内,以速度35rpm进行磁力搅拌,加入30ml石油醚,以2℃/min的速度进行加热并持续搅拌,向瓶内通入氮气,通入速度为3l/min,瓶口以滤纸盖封,同时加热至55℃,溶液逐渐澄清后出现浑浊时,以2℃/min左右的速率来降低温度直至室温;保持室温,搅拌速度和氮气通入速率,当目测母液减少至原体积的1/3左右(约75-77ml)时,停止搅拌,除去瓶口滤纸,补入水,至溶剂为原体积50-55%,继续以35rpm速度搅拌,以4l/min速率通入氮气,以2℃/min速度加热,加热至60℃,当溶剂逐渐浓缩至原母液体积40%时,停止加热,以1℃/min的速度降温至5℃后停止搅拌,停止通入氮气,静置析晶过夜(12h);固液过滤分离,将沉淀分散置于三层网带烘干桶上,以80℃热风加热30min后即得。
34.检测纯度99.6%(hplc),单杂含量<0.02%,检测图谱和实施例1相同。为单晶结构。
35.实施例4
36.将100g盐酸多巴酚丁胺粗品加入盛有200ml无水乙醇的反应瓶中,置于通风橱内,以速度35rpm进行磁力搅拌,加入20ml石油醚,以2℃/min的速度进行加热并持续搅拌,向瓶内通入氮气,通入速度为3l/min,瓶口以滤纸盖封,同时加热至55℃,溶液逐渐澄清后出现浑浊时,增加搅拌速度至60rpm,氮气通入速度增加至4l/min,逐渐以1℃/min左右的速率来降低温度直至室温;保持室温,搅拌速度和氮气通入速率,约2-4h后目测母液减少至原体积的1/4(约55ml)时,停止搅拌,除去瓶口滤纸,补入无水乙醇55ml,至溶剂为原体积50%(110ml),继续以60rpm速度搅拌,以4l/min速率通入氮气,以2℃/min速度加热,加热至50℃,当目测母液减少至原体积的1/3左右时,停止搅拌,除去瓶口滤纸,补入水,至溶剂为原体积50-55%,继续以60rpm速度搅拌,以4l/min速率通入氮气,以2℃/min速度加热,加热至60℃,当溶剂逐渐浓缩至原母液体积40%时,停止加热,以1℃/min的速度降温至5℃后停止搅拌,停止通入氮气,静置析晶过夜(18h);固液分离,将沉淀90℃真空干燥,真空度1-0.97pa。
37.检测纯度99.7%(hplc),单杂含量<0.02%,检测图谱和实施例1相同,单晶结构。
38.实施例5
39..将100g盐酸多巴酚丁胺粗品加入盛有200ml无水乙醇的反应瓶中,置于通风橱内,以速度35rpm进行磁力搅拌,以2℃/min的速度进行加热并持续搅拌,加入20ml丙酮,向瓶内通入氮气,通入速度为3l/min,瓶口以滤纸盖封,同时加热至55℃,增加搅拌速度至60rpm,氮气通入速度增加至4l/min,逐渐以1℃/min左右的速率来降低温度直至室温;保持室温,搅拌速度和氮气通入速率,约4-8h后目测母液减少至原体积的1/3左右(时,停止搅拌,除去瓶口滤纸,补入水,至溶剂为原体积50-55%,继续以60rpm速度搅拌,以4l/min速率通入氮气,以2℃/min速度加热,加热至60℃,当溶剂逐渐浓缩至原母液体积40%时,停止加热,以1℃/min的速度降温至5℃后停止搅拌,停止通入氮气,静置析晶过夜(18h);固液分离,将沉淀分散置于三层网带烘干桶上,以90℃热风加热15min后即得。
40.检测纯度97.7%(hplc),单杂含量<0.05%,xrd衍射多晶结构。
41.实施例6
42.将100g盐酸多巴酚丁胺粗品加入盛有200ml无水乙醇的反应瓶中,置于通风橱内,以速度50rpm进行磁力搅拌,加入20ml石油醚,以2℃/min的速度进行加热并持续搅拌,向瓶内通入氮气,通入速度为3l/min,瓶口以滤纸盖封,同时加热至35-55℃,溶液逐渐澄清后出现浑浊时自然降温直至室温;保持室温,搅拌速度和氮气通入速率,约2-4h后目测母液减少至原体积的1/3左右时,停止搅拌,除去瓶口滤纸,补入水,至溶剂为原体积50-55%,继续以50rpm速度搅拌,以2l/min速率通入氮气,以2℃/min速度加热,加热至60℃,当溶剂逐渐浓缩至原母液体积40%时,停止加热,以1℃/min的速度降温至5℃后停止搅拌,停止通入氮气,静置析晶过夜(18h);固液过滤分离,将沉淀分散置于三层网带烘干桶上,以105℃热风加热50min后即得。
43.检测纯度99.3%(hplc),单杂含量<0.01%,检测图谱和实施例1相同。单晶结构,
44.实施例7
45.将100g盐酸多巴酚丁胺粗品加入盛有200ml无水乙醇的反应瓶中,置于通风橱内,以速度50rpm进行磁力搅拌,加入20ml石油醚,加热并持续搅拌,向瓶内通入氮气,通入速度
为3l/min,瓶口以滤纸盖封,加热至55℃,溶液逐渐澄清后出现浑浊时,增加搅拌速度至60rpm,氮气通入速度增加至4l/min,逐渐以1℃/min左右的速率来降低温度直至室温;保持室温,搅拌速度和氮气通入速率,约2-4h后目测母液减少至原体积的1/3左右时,停止搅拌,除去瓶口滤纸,继续以60rpm速度搅拌,以4l/min速率通入氮气,以2℃/min速度加热,加热至60℃,当溶剂逐渐浓缩至原母液体积10%时,停止加热,以1℃/min的速度降温至5℃后停止搅拌,停止通入氮气,密封瓶口,静置析晶过夜(18h);固液过滤分离,将沉淀分散置于三层网带烘干桶上,以80℃热风加热10min后即得。
46.检测纯度98.7%(hplc),单杂含量<0.03%,xrd衍射显示为多晶结构。
47.实施例8
48.将100g盐酸多巴酚丁胺粗品加入盛有300ml乙醇的反应瓶中,置于通风橱内,以速度40rpm进行磁力搅拌,加入30ml石油醚,以2℃/min的速度进行加热并持续搅拌,向瓶内通入氮气,通入速度为1l/min,瓶口以滤纸盖封,同时加热至55℃,溶液逐渐澄清后出现浑浊时,增加搅拌速度至60rpm,氮气通入速度增加至4l/min,逐渐以1℃/min左右的速率来降低温度直至室温(24℃);保持室温,搅拌速度和氮气通入速率,约2-4h后目测母液减少至原体积的40%时,停止搅拌,除去瓶口滤纸,补入水,至溶剂为原体积50-55%,继续以60rpm速度搅拌,以4l/min速率通入氮气,当溶剂逐渐浓缩至原母液体积40%时,停止搅拌,停止通入氮气,室温下静置析晶过夜(18h);固液过滤分离,将沉淀分散置于三层网带烘干桶上,以90℃热风加热10min后即得。
49.检测纯度99.1%(hplc),单杂含量<0.01%,xrd衍射显示为多晶型。
50.实施例9
51.将100g盐酸多巴酚丁胺粗品加入盛有300ml乙醇的反应瓶中,置于通风橱内,以速度40rpm进行磁力搅拌,加入25ml石油醚,以2℃/min的速度进行加热并持续搅拌,向瓶内通入氮气,通入速度为3l/min,瓶口以滤纸盖封,同时加热至35-55℃,溶液逐渐澄清后出现浑浊时,增加搅拌速度至70rpm,氮气通入速度增加至4l/min,自然降温直至室温(24℃);保持室温,保持搅拌速度和氮气通入速率,约2-4h后目测母液减少至原体积的30%左右时,停止搅拌,除去瓶口滤纸,补入水,至溶剂为原体积50%,继续以70rpm速度搅拌,以4l/min速率通入氮气,以2℃/min速度加热,加热至60℃,当溶剂逐渐浓缩至原母液体积40%时,停止加热,以1℃/min的速度降温至9℃后停止搅拌,停止通入氮气,静置析晶过夜(18h);固液过滤分离,将沉淀分散置于三层网带烘干桶上,以75℃热风加热10min后即得。
52.检测纯度99.23%(hplc),单杂含量<0.2%。检测图谱和实施例1相同,单晶结构。
53.对照例1
54.将50g盐酸多巴酚丁胺粗品加入50g甲醇中,加热至60℃揽拌溶解,再缓慢加入100g纯化水,停止加热,自然冷却至室溫,放入4℃冰箱静置12h,减压过滤,滤饼放入真空干燥箱55℃干燥化,即得。
55.对照例2
56.将乙醇替换为含有少量盐酸的水溶液(0.1mmol/l),其他步骤和实施例1相同,最终产品纯度96.8%,单杂含量>0.5%,不符合原料药标准,多晶型。
57.对照例3
58.步骤2-3中不进行加热,其余步骤和实施例2相同,无结晶形成,仅有无定型沉淀。
59.原研药理化性能试验:(结果参见表1)
60.1)粒径分布检测,取上述实施例产品,置于pss纳米激光粒度仪样品槽中,激光粒度仪软件采集数据,使用电子探针显微分析仪放大观察药颗粒晶体形态。
61.2)吸湿性测定,使用水蒸气吸附分析仪评价各实施例产物在室温条件下的吸湿性,在相对湿度80%进行测试,计算其在储存3个月后的的吸湿增重情况,(以吸湿增重的质量百分数表示)。
62.3)xrd衍射确定产品单晶/多晶型或无定型状态。
63.4)色差确定,在暗室中将待测品置于白色桌面,恒温恒湿(20℃,50%湿度)条件;先使用校正后的色差仪进行检测,产物干燥后即刻检测作为参照标准,12h后再次检测,计算δe值。
64.表1
[0065][0066][0067]
结晶过程对温度和轻微的环境变化非常敏感,现有技术结晶虽然提高纯度,但通常无法作为原研药用于制剂生产,其中的颗粒大小,形态,均匀度,光敏性等存在诸多操作问题,均需要在制剂中加入辅料或进一步粉碎加工;而通常情况下,剧烈的过程会导致杂质增加和结晶难于形成;本发明在纯化中大量通入氮气的同时促进溶剂挥发,使得药物制备过程中的有机杂质去除较为彻底,同时实施例1,9的粒径明显小于其他实施例,并且正态分布较好,实施例2-8的产品粒径(d90)取值较大也有部分原因是颗粒大小不均导致,特别是实施例2、8的颗粒粒径分布方差较大,无法作为原研药原料直接用于制剂,配置液体制剂时难于溶解完全,可能与溶剂挥发速率,搅拌或干燥过程有关,需要进一步分析制备过程中的工艺差异导致晶体结构差异的原因;由上述试验结果也可看出,溶剂使用不当会导致单晶无法形成,本发明发现了几个关键的制备要素,例如温度变化,补充母液时机和干燥工艺,其有利于原药晶体的形成。
[0068]
上述部分实施例产品显示了较高的理化性能,特别是色差值较小,粒径小,在短时间能颜色变化几乎难于察觉,晶体较为紧实,形态也更为规则均匀,适用于临床快速配置药物应用。在吸湿性上也显示出良好性能,而部分实施例晶体具有高吸湿性,说明降温速度过快或挥发溶剂的过程可影响晶体的结构性能,在析晶过程中(晶核形成后),环境变化特别
是温度变化需要变缓,而真空干燥可能剧烈的溶剂挥发的同时对晶体的结构也有一定影响,导致吸湿性也高;同时,颗粒均匀度较差(而非粒径大)的产品在储存过程中似乎也更容易结块。
[0069]
上述试验仅仅是选择了具有代表性的关键步骤的若干实验例。上述具体实施例并不构成对本发明的保护范围的限定,本领域技术人员可以根据上述说明对本发明进行各种变化和应用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1