一种基于平衡基因表达高效合成核黄素的方法
1.本发明涉及一种基于平衡基因表达高效合成核黄素的方法,具体涉及mrna工程平衡操纵子内基因表达高效合成核黄素的策略,属于合成生物学技术领域。
背景技术:
2.核黄素(vb2)是人类和动物必需的营养素,广泛用于制药、食品添加剂和化妆品。枯草芽孢杆菌(bacillus subtilis)是一种革兰氏阳性细菌,通过“经典”菌株改良策略具有优异的核黄素生产性能。前体供应被认为是核黄素生物合成的主要限制因素。在枯草杆菌中,葡萄糖-6-磷酸酶脱氢酶(由zwf编码)和6-磷酸葡萄糖酸脱氢酶(由gnd编码)通过磷酸戊糖(pp)途径将葡萄糖-6-磷酸转化为核酮糖-5-磷酸。核黄素是由直接前体5-磷酸核糖和三磷酸鸟苷(gtp)经核黄素合成途径,由rib操纵子编码的一系列酶催化合成的。基因ribba编码一种双功能gtp环水解酶ii/3,4-二羟基-2-丁酮4-磷酸合酶,该合酶催化从核酮糖-5-磷酸和2,5-二氨基-6-核糖氨基-4(3h)-嘧啶酮-50-磷酸(darpp)合成3,4-二羟基-2-丁酮4-磷酸(dhpb)。增加基因ribba编码的酶的活性可使核黄素的效价增加25%。磷酸核糖焦磷酸盐(prpp)是细胞中通过从头嘌呤途径形成的gtp前体,首先转化为肌苷单磷酸(imp),然后imp随后转化为gtp。谷氨酸棒杆菌突变基因zwf243和gnd361的联合过表达使补料分批发酵中的核黄素产量增加了39%。通过在培养基中添加额外的gtp,并通过代谢工程策略将代谢重新定向到嘌呤途径,核黄素产量显著增加。因此,gtp是核黄素合成的主要限制因素。通过代谢工程策略消除反馈抑制,核黄素产量增加了3倍。通过增加pp途径的代谢流量和增加细胞内嘌呤途径前体prpp的供应,可以显著提高核黄素的产量。
3.基因缺失和过度表达已成为传统代谢工程和基因功能研究的主要策略。在大肠杆菌中,过度表达和敲除技术用于上调或下调基因表达,以促进天然和非天然产物。将天然和非天然产物代谢途径引入工程菌株合成化合物时,往往需要多种酶同时参与。微调多酶催化反应系统中的蛋白质丰度,使细胞达到最佳生长和生产状态。最近的研究使用了几种策略来控制细胞内蛋白质丰度,例如启动子文库、rbs文库、5'-utr文库、mrna稳定性和crispri系统。这些策略用于微调基因表达水平,并控制多个基因的协调表达,以优化代谢流量。启动子是合成生物学中的关键调控元件,控制基因表达的强度和时间。通过启动子工程微调该途径的基因表达,以平衡代谢流量。为了平衡平行的多基因表达水平的平行性,一种快速且模块化的方法将基因与一组紧密的核糖体结合位点配对,这些位点可以将蛋白质丰度调节几个数量级。emopec(蛋白质表达变化的经验模型和寡核苷酸)是一种基于sd(shine dalgarno)序列对蛋白质表达的贡献而开发的工具,可以通过改变一些碱基来调节任何大肠杆菌基因的表达水平。此外,通过生成可调基因间区文库(tigr)、重组各种转录后控制元件和筛选所需的相对表达水平来协调操纵子中多个基因的表达,用于合成非天然产物。这种方法可以通过基因间的协同表达有效减少有毒中间代谢物的积累和蛋白质表达的冗余。
4.在代谢工程实践中,前体供应不足是限制目标产品产量的主要瓶颈。高水平表达
限速酶以驱动目标产物生物合成途径的代谢流量是增加前体合成的快速而直接的策略。复杂化合物,尤其是天然产物,通常由多种前体通过一系列酶促反应进行催化。例如,阿片类物质蒂巴因和氢可酮的生物合成途径包含21和23种来自植物、哺乳动物、酵母和细菌的酶反应。细胞中前体的比例直接影响化合物的合成效率。浓度最低的前体将成为限制因素,而浓度较高的前体可能对细胞有毒。在生产过程中,基因表达必须适当,以避免速率限制和有毒前体的积累。基于合成生物学的各种策略已被开发用于平衡细胞内代谢,包括启动子工程、rbs文库和动态控制。
5.在枯草芽孢杆菌合成核黄素的过程中,前体供应不足是限制进一步提高核黄素产量的主要因素,目前单纯的通过表达和敲除代谢途径关键基因虽然可以促进核黄素的合成,并不能使细胞内平衡代谢流的平衡,而细胞内代谢流分配失衡导致副产物增多,造成资源浪费的现象。
技术实现要素:
6.本发明提供一种能够高效有效调节细胞内代谢平衡的策略,并用此策略构建高产核黄素的工程菌株,并且采用该基因工程菌生产核黄素,解除发酵过程中的代谢流不平衡限制,提高核黄素产量。设计了一个基因间可调区域库(tigrs)来调节合成操纵子内多个基因的表达。tigrs文库由mrna的二级结构、rna酶切割位点和rbs序列组成,并用来控制mrna的加工和稳定。操纵子内基因转录成完整的mrna链,通过细胞内的rnase e(来源于大肠杆菌基因rne编码)识别tigr序列上的特殊位点,将mrna分割成单个转录单元,同时每个转录单元形成独特的5’和3’结构,通过影响mrna的稳定性和rbs的结构,进而影响蛋白水平。
7.本发明提供了一种调节基因表达量的元件,所述元件由mrna二级结构、rnase e切割位点和rbs序列组成,所述元件的核苷酸序列如seq id no.7所示。
8.本发明提供了一种调控基因表达的方法,利用所述的元件调控基因的表达,所述基因为两个或多个;
9.所述方法为:
10.(1)利用权利要求1所述的元件将基因连接,并整合至表达载体上构建得到重组质粒;
11.(2)将重组质粒转入大肠杆菌中,利用表达载体上的抗性基因筛选阳性转化子;
12.(3)从阳性转化子中提取质粒,转入整合了rnase e基因的宿主细胞中得到重组细胞,将重组细胞进行培养并检测荧光强度,根据荧光强度与基因表达强度成正相关筛选对应的重组细胞。
13.本发明提供了一种生产核黄素的基因工程菌,利用所述表达元件调控zwf、ribba和ywlf基因的表达,所述基因工程菌的基因组上还整合了来源于大肠杆菌的rnase e基因。
14.在一种实施方式中,所述基因zwf、ribba和ywlf的核苷酸序列分别如seq id no.20~22所示
15.在一种实施方式中,所述zwf基因和ribba基因之间由seq id no.10、12、14、16或18连接;所述ribba和ywlf基因之间由seq id no.11、13、15、17或18连接。
16.优选地,利用seq id no.14和seq id no.15分别连接zwf基因和ribba基因、ribba和ywlf基因。
17.在有一种实施方式中,利用质粒pma5-sat为表达载体,所述质粒pma5-sat记载于公开号为cn104531745a的专利文献中。
18.在一种实施方式中,所述基因工程菌以枯草芽孢杆菌rf1为宿主细胞,所述枯草芽孢杆菌rf1公开于公开号为cn104531745a的专利文献中。
19.本发明提供了一种生产核黄素的方法,是利用生产核黄素的基因工程菌发酵生产核黄素。
20.在一种实施方式中,将所述基因工程菌培养至od
600
=24~26以3%(v/v)的量添加至摇瓶发酵体系中,在37℃~45℃、150~250rpm下发酵生产不少于24h。
21.在一种实施方式中,所述摇瓶发酵体系中含有15~25g/l葡萄糖,15~25g/l酵母粉,2~5g/l柠檬酸铵,0.5~2g/l k2hpo4,0.5~2g/l kh2po4,1~5g/l mgso4·
7h2o,0.01~0.05g/l mncl2,0.01~0.1g/l cacl2,1~5g/l cuso4,ph 6.8。
22.在一种实施方式中,将所述基因工程菌培养至od
600
=20~25以3%(v/v)的量添加至分批补料发酵体系中,并添加补料培养基,以保持葡萄糖浓度不低于5g/l,在37℃~45℃、400~800rpm下发酵生产不少于24h。
23.在一种实施方式中,分批补料发酵体系中含有15~25g/l葡萄糖,15~25g/l酵母粉,2~9g/l(nh4)2hpo4,3~8g/l k2hpo4,1~1.5g/l mgso4·
7h2o,0.01~0.05g/l znso4·
7h2o,0.01~0.05g/l mncl2,0.01~0.02g/l feso4·
7h2o。
24.在一种实施方式中,补料培养基中含有500~600g/l葡萄糖,5~10g/l酵母粉,4~6g/l(nh4)2hpo4,1~5g/l k2hpo4,0.1~1g/l mgso4·
7h2o。
25.本发明提供了所述基因工程菌在生产核黄素及其衍生物中的应用。
26.有益效果:
27.(1)使用双荧光报告基因gfp和mcherry表征tigr序列的调节能力,在枯草芽孢杆菌中,操纵子中第二个基因的表达明显高于第一个基因的表达,红色和绿色的相对荧光比变化超过70倍(从4:1mcherry/egfp到18:1egfp/mcherry)。在tigr文库中,mcherry的荧光变化范围超过80倍,egfp的荧光变化范围超过40倍。使用tigr文库生成一系列包含zwf、ribba和ywlf基因的操纵子,并筛选最优的操纵子组合。从5000个多克隆的文库筛选到5株高产核黄素的工程菌。
28.(2)在摇瓶发酵水平,筛选到的工程菌rf1-l3的核黄素效价达到2.7g/l,与亲本相比增加了64.35%。
29.(3)在5-l发酵水平,与亲本菌株rf1相比,工程菌株rf1-l3的核黄素滴度在48小时时增加59.27%,达到11.77g/l。
附图说明
30.图1为tigr序列结构示意图;
31.图2为tigr报告基因文库示意图;
32.图3为tigr文库在枯草芽孢杆菌中调控能力分析;
33.图4为摇瓶发酵分析高产核黄素菌株合成核黄素能力;
34.图5为5l发酵罐分析rf1和rf-l3合成核黄素能力。
具体实施方式
35.下述实施例中所涉及的培养基如下:
36.种子培养基:40g/l葡萄糖、5g/l酵母膏、10g/l蛋白胨、10g/l nacl和10μg/m l氯霉素。
37.摇瓶发酵培养基:20g/l葡萄糖,20g/l酵母粉,4g/l柠檬酸铵,1g/l k2hpo4,1g/l kh2po4,2g/l mgso4·
7h2o,0.04g/l mncl2,0.06g/l cacl2,2g/l cuso4,ph 6.8。
38.分批补料发酵培养基:20g/l葡萄糖,20g/l酵母粉,6g/l(nh4)2hpo4,5g/l k2hpo4,1.5g/l mgso4·
7h2o,0.03g/l znso4·
7h2o,0.05g/l mncl2,0.02g/l feso4·
7h2o。
39.补料培养基:600g/l葡萄糖,10g/l酵母粉,6g/l(nh4)2hpo4,5g/l k2hpo4,0.5g/l mgso4·
7h2o。
40.最低培养基含有葡萄糖:20.0g/l、(nh4)2so
4 2.0g/l、kh2po
4 13.1g/l、k2hpo
4 6.0g/l、nac6h5o7·
2h2o 1.2g/l、mgso4·
7h2o 0.05g/l,并补充色氨酸、苯丙氨酸和酪氨酸(各25mg/l)。
41.下述实施例中所涉及的检测方法如下:
42.测定egfp和mcherry的荧光值,以评估pmtg文库的表达范围。将含有pmtg文库的大肠杆菌和枯草杆菌接种在含有200μl lb培养基的无菌96个黑孔板(康宁3603)中,并在37℃下培养10小时。此外,egfp荧光(激发,490nm;发射,530nm)、mcherry荧光(激发,588nm;发射,633nm),光密度(600nm处的吸光度)在培养结束时用微孔板多模式读取器(biotek,cytation 3)测定。使用方程式(1)计算相对荧光密度。fpbg表示不含荧光蛋白的菌株的荧光值,odbg表示培养基的吸光度。
[0043][0044]
核黄素在440~500nm波长的光照射下发出黄绿色荧光,其荧光强度与其在稀溶液中的浓度成正比。对于核黄素标准曲线,将10mm核黄素母液稀释至不同浓度,包括10mm、5mm、2.5mm、1.25mm、0.625mm、0.313mm和0.1562mm。在激发波长444nm和吸收波长500nm处测量荧光值,并绘制标准曲线。将来自工程菌株库的克隆接种到含有200μl最低培养基的96孔板中,在37℃下以220rpm的转速振荡培养24小时。使用0.01mnaoh将细胞培养物稀释至适当浓度,并通过微孔板多模式读取器(biotek,cytation 3)测量荧光强度。用分光光度计监测od
600
nm下细胞的生长。
[0045]
将制备得到的发酵液用0.01m naoh稀释,然后在12000rpm下离心2min,取上清液测定核黄素浓度。将上清液转移到新的ep管中,并稀释至合适的浓度范围(0.3-0.8),使用分光光度计在od
444
nm处测量吸光度值。核黄素浓度根据核黄素浓度标准曲线计算。按照核黄素标准曲线计算公式为:od
444
*稀释倍数*30/1000。
[0046]
使用glucose analysis(model-sba40,shandong,china)测量葡萄糖浓度。
[0047]
下述实施例中所使用的对照菌株rf1-apagatgv、rf1均公开于公开号为cn104531745a的专利文献中。
[0048]
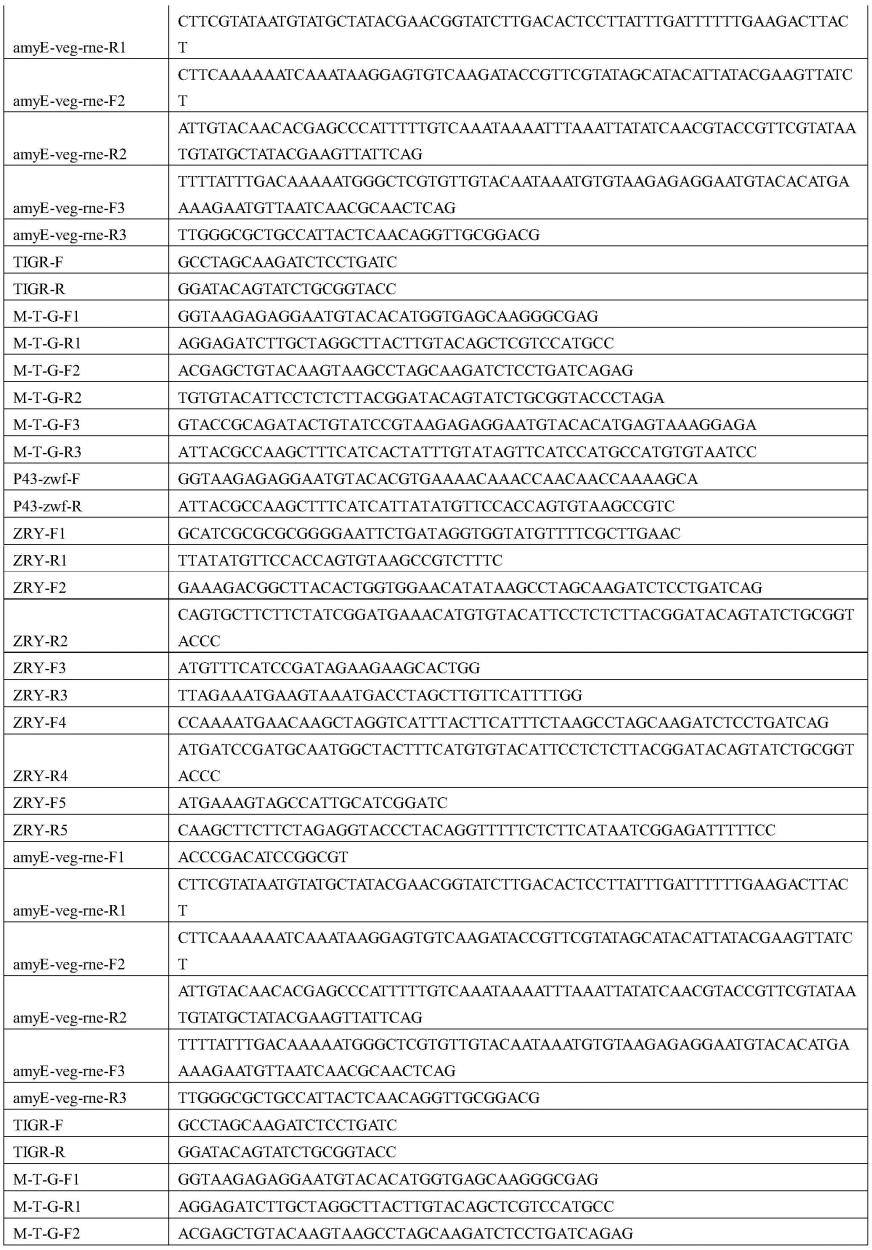
下述实施例中所涉及的引物序列如表1所示:
[0049]
表1引物序列
[0050][0051][0052][0053]
实施例1:在枯草芽孢杆菌中异源表达基因rne
[0054]
利用同源重组的方式将来源于大肠杆菌的rnase e基因rne整合到枯草芽孢杆菌
168的基因组上的基因amye(gene id:938356)处。
[0055]
具体步骤如下:
[0056]
(1)按照表1的引物序列扩增基因amye上游同源臂(1000bp)和下游同源臂(1000bp),分别获得amye上游、下游同源臂片段;并通过琼脂糖凝胶电泳分离pcr产物,并切胶回收目的pcr产物。然后将上下游同源臂、pveg-rne片段(片段核苷酸序列如seq id no.1所示)与抗性marker(博来霉素抗性基因,核苷酸序列如seq id no.2所示)通过融合pcr的策略进行融合。
[0057]
首先将上下游同源臂、pveg-rne片段与抗性marker片段按体积比1:1混合,加入等体积的pcr酶进行融合pcr反应(条件为98℃3min,98℃8s,61℃5s,72℃2min,扩增13个循环),以该步反应后的产物为模板,使用引物amye-veg-rne-f1和amye-veg-rne-r3扩增融合片段(反应条件为:98℃3min,98℃10s,58℃15s,72℃1min,扩增34个循环)。将pcr产物纯化回收用于敲除反应,融合后的片段含有博来霉素抗性基因和lox66-lox71重组位点,便于后期抗性marker的消除。
[0058]
(2)将步骤(1)得到的融合pcr产物利用化转的方法化转至bacillus subtilis168感受态细胞中,并涂布在含有博来霉素抗性的lb平板,37℃培养12h,抗性平板上生长的菌株使用菌落pcr方法验证整合是否成功,验证引物使用amye-veg-rne-f1和amye-veg-rne-r3,经pcr验证之后得到整合成功的阳性转化子。
[0059]
(3)将pdg148质粒化转至步骤(2)制备得到的正确的阳性转化子中,在180rpm、37℃震荡培养24h,然后取部分培养液涂布于lb平板上,37℃培养12h至长出单菌落,用无菌牙签将lb平板上的单菌落一一对应点到另一块含有博来霉素的抗性平板上,在37℃培养培养12h,在lb平板上可以生长且在博来霉素抗性平板上无法生长的菌落为消除博来霉素抗性的敲除菌株。
[0060]
将得到的敲除菌株在180rpm、42℃震荡培养24h用来消除pdg148质粒。将部分培养液涂布于lb固体培养基上,然后37℃培养培养12h后,用无菌牙签将lb平板上的菌落一一对应点到另一块含有氨苄青霉素的抗性平板上,37℃培养培养12h,在lb平板上可以生长,而在博来霉素抗性平板上无法生长的菌落为消除质pdg148粒的敲除菌株。最后得到的菌株为无痕敲除的目的菌株bsf01。
[0061]
实施例2:含有双荧光报告基因的tigr文库构建
[0062]
tigrs文库包括mrna二级结构、rnase e切割位点和rbs序列控制元件。tigrs文库利用mrna的二级结构、rnase e酶切割位点和rbs序列控制mrna的加工和稳定,从而影响操纵子上基因的表达(图1)。
[0063]
为了进一步扩增适用于枯草杆菌的合成生物工具,基于tigr构建m-tigr-g文库(图2),并将tigrs文库导入菌株bsf01,该菌株在amye位点过表达来源于大肠杆菌的基因rne。测定两个报告基因的荧光值,以评估枯草杆菌中tigr文库的调节能力。
[0064]
具体步骤如下:
[0065]
1、tigr文库的制备
[0066]
(1)使用pcr合成tigr,将寡核苷酸序列tigr-a、tigr-b、tigr-c和tigr-d(核苷酸序列分别如seq id no.3~6所示)组装成嵌合dna序列(核苷酸序列如seq id no.7所示)。将每个寡核苷酸序列中的五个核苷酸替换为随机核苷酸“n”,以合成多样性序列。通过融合
pcr整合mcherry基因、tigr序列(嵌合dna序列)和gfp基因构建融合片段m-tigrs-g。然后,通过gibson组装将融合pcr片段m-tigrs-g插入质粒pp43nmk中,构建用于筛选tigr的报告质粒pmtg,使得报告操纵子的转录由强启动子p43控制。将质粒pmtg转化到有活性的大肠杆菌jm109细胞中,并用氨苄青霉素筛选转化子。刮取大肠杆菌文库菌落并提取质粒,从三个单独的平板上收集,并转化到bsf01中。
[0067]
(2)将步骤(1)得到的多克隆文库接种到96孔板中,测定gfp和mcherry荧光值,构建tigr文库。
[0068]
结果显示:操纵子中第二个基因的表达明显高于第一个基因的表达,根据基因间区域的不同,红色和绿色的相对荧光比变化超过70倍(从mcherry/egfp=4:1到egfp/mcherry=18:1;图3)。
[0069]
实施例3:构建tigr文库筛选高产核黄素工程菌株
[0070]
按照实施例1的方式,在核黄素生产菌株rf1的基因amye处整合来源于大肠杆菌的rnase e基因rne,构建得到菌株rf1-r。
[0071]
使用tigr文库来产生一系列包含zwf、ribba和ywlf基因的合成操纵子,这些操纵子随后筛选出增加核黄素产量的最优组合。使用巨引物pcr方法,在操纵子的第一个和第二个基因以及第二个和第三个基因之间同时引入tigr文库。基因zwf被用作操纵子的第一个基因,ribba和ywlf被用作操纵子的第二和第三个基因。
[0072]
具体步骤如下:
[0073]
(1)通过融合pcr构建含有tigr文库的基因片段。首先,设计引物zry-f2/zry-r2、zry-f4/zry-r4,以实施例2中的tigr文库(核苷酸序列如seq id no.7所示的嵌合dna序列)为模板,扩增基因zwf、ribba和ywlf之间的tigr文库。将pcr片段zwf、tigr文库进行融合pcr,获得zwf-tigr(核苷酸序列如seq id no.8所示)。根据上述方法获得了ribba-tigr的pcr片段(核苷酸序列如seq id no.9所示)。将质粒pma5-sat使用限制性内切酶ecori和kpni在37℃处理30min并纯化回收,然后将纯化的pcr片段zwf-tigr、ribba-tigr和ywlf通过融合pcr方法连接成一条pcr片段,并插入质粒pma5-sat,使用gibson组装获得zwf-tigr-ribba-tigr-ywlf文库,并转化到大肠杆菌dh5α感受态中,37℃培养16h,并通过菌落pcr筛选正确转化子。将得到的转化子混合提取质粒得到文库并转化到rf1-r中构建工程菌株文库,将来自工程菌株库的克隆接种到含有200μl最低培养基的96孔板中,在37℃下以220rpm的转速振荡培养24小时。使用0.01m naoh将细胞培养物稀释至适当浓度,并通过微孔板多模式读取器(biotek,cytation 3)测量荧光强度,筛选荧光强度最高的突变株。
[0074]
实施例4:摇瓶阶段采用基因工程菌株发酵生产核黄素
[0075]
使用摇瓶发酵对实施例3中筛选得到的荧光强度最高的突变株菌株进行复筛。
[0076]
将单个菌落接种在10ml-lb培养基中,并在37℃下以180rpm的转速摇动培养。16小时后,按照1%(v/v)接种量将培养物转移至含有50ml lbg培养基(40g/l葡萄糖、5g/l酵母提取物、10g/l蛋白胨和10g/l nacl)的250ml挡板摇瓶中,并在41℃下以220rpm的转速振荡24小时得到种子培养物。种子培养物(od
600
=25.21)转移至含有50ml发酵培养基(3%(v/v)接种量)的500ml挡板摇瓶中,然后在41℃下以220rpm的转速振荡培养48小时。在摇瓶发酵期间,每12小时取样1毫升细胞悬浮液,以测量od
600
和核黄素浓度。
[0077]
结果表明,五株工程菌的核黄素效价均显著提高,其中以工程菌rf1-l3的效价优
势最为明显。工程菌rf1-l3的核黄素效价达到2.7g/l,比亲本的1.67g/l提高了64.35%(图4为发酵48h结束后各菌株的核黄素产量),工程菌rf1-l1、rf1-l2、rf1-l4、rf1-l5的核黄素效价分别为1.94g/l、2.01g/l、2.16g/l、2.24g/l。
[0078]
表2菌株rf1-l1~rf1-l5的tigr序列
[0079][0080]
[0081][0082]
实施例5:发酵罐水平采用基因工程菌株发酵生产核黄素
[0083]
具体步骤如下:
[0084]
(1)分别将在10ml lb培养基中培养24h的枯草芽孢杆菌菌株rf1-apagatgv、rf1按照体积比3%(v/v)的接种量接种到100ml种子培养基中(种子培养基包括20g/l葡萄糖,20g/l酵母粉,4g/l柠檬酸铵,1g/l k2hpo4,1g/l kh2po4,,2g/l mgso4·
7h2o,0.04g/l mncl2,0.06g/l cacl2,2g/l cuso4),温度为41℃,转速为180rpm,培养16h后,制备得到种子液(od
600
=23.5);
[0085]
(2)将制备得到的100ml种子液全部接种到含有1900ml的发酵培养基的5l发酵罐中,进行分批补料发酵。
[0086]
通过控制补料培养基流量,使发酵液中的剩余葡萄糖浓度保持在不低于5g/l。发酵过程中,使用1m h2so4和50%氨水保持发酵液ph为6.8。在开始分批进料前,转速保持在400rpm,然后逐渐将转速提高到900rpm直到发酵结束,温度始终保持在41℃。
[0087]
结果显示:在5l生物反应器中进行补料分批发酵,以验证工程菌枯草杆菌rf1-l3在核黄素生产中的大规模发酵性能。在补料分批发酵中,工程菌和亲本菌株的生长没有明显差异。与亲本菌株rf1(7.39g/l)及rf1-apagatgv(10.71g/l)相比,工程菌株rf1-l3的核黄素滴度在48h时增加了59.27%,达到11.77g/l(图5a和图5b)。
[0088]
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1