一种人源ATP合成酶的纯化方法
一种人源atp合成酶的纯化方法
技术领域
1.本发明属于蛋白纯化技术领域,具体涉及一种人源atp合成酶的纯化方法。
背景技术:
2.atp合成酶是生物体合成能量货币atp的蛋白复合物,是氧化磷酸化系统最后也是最重要的一员,能够将电子传递链复合物形成的质子梯度转化为化学能储存在atp中。由于atp合成酶在生物体正常功能发挥中的重要作用,atp合成酶异常往往与疾病密切相关,比如神经退行性疾病、肿瘤等。atp合成酶许多位点的突变会导致不同的癌症或其他疾病。此外,鉴于肿瘤细胞增殖对于氧化磷酸化通路能量供给的需求,越来越多的靶向atp合成酶的新型抗肿瘤药物被开发出来,并表现出了良好的抗肿瘤活性。
3.近年来,哺乳动物atp合成酶的结构和功能研究有了重大进展,但是至今无人纯化出纯度高、有活性的人源atp合成酶并解析结构,当前用于研究线粒体蛋白主流的密度梯度离心的方法对设备要求较高,超速离心机价格昂贵,通常需要离心24h,离心时间长,并且纯度不高,使我们对人源atp合成酶的结构功能机制的了解仍有限,由此制约了针对该靶点的新药开发。研究人源atp合成酶的结构与功能,对于深入了解人体细胞atp的合成机制、突变致病机制及药物发现等都具有的意义。
技术实现要素:
4.本发明的目的在于提供一种人源atp合成酶的纯化方法,获得高纯度人源atp合成酶,降低生产成本。
5.本发明提供了一种人源atp合成酶的纯化方法,包括如下步骤:
6.制备人源线粒体粗提物,溶膜,得线粒体总蛋白溶液;
7.利用阳离子交换柱对所述线粒体总蛋白溶液进行第一洗脱,收集洗脱液,得第一纯化液;
8.利用阴离子交换柱对所述第一纯化液进行第二洗脱,收集洗脱液,得第二纯化液;
9.利用凝胶排阻色谱柱对所述第二纯化液进行第三洗脱,得纯化的人源atp合成酶。
10.优选的,所述溶膜的时间为30min~120min。
11.优选的,所述溶膜的方式包括加入去污剂;所述去污剂包括lmng、digitonin和gdn中的一种或多种。
12.优选的,利用缓冲液c和缓冲液d进行所述第一洗脱;
13.所述缓冲液c包括20mm 2-(n-吗啡啉)乙磺酸和0.004%w/v去污剂;所述缓冲液c的ph为6.0;
14.所述缓冲液d包括20mm 2-(n-吗啡啉)乙磺酸、1m nacl和0.004%w/v去污剂;所述缓冲液d的ph为6.0。
15.优选的,所述第一洗脱的流速为1ml/min;所述第一洗脱的方式为梯度洗脱;
16.所述梯度洗脱的程序为:
17.0min:所述缓冲液c的体积百分含量为95%;所述缓冲液d的体积百分含量为5%;
18.0~40min:所述缓冲液c的体积百分含量由95%匀速减少到75%;所述缓冲液d的体积百分含量由5%匀速增加到25%;
19.优选的,依次利用缓冲液f和缓冲液g进行所述第二洗脱;
20.所述缓冲液f包括20mm mops和0.004%w/v去污剂;所述缓冲液f的ph为7.4;
21.所述缓冲液g包括20mm mops、1m nacl和0.004%w/v去污剂;所述缓冲液d的ph为7.4。
22.优选的,所述第二洗脱的流速为1ml/min;所述第二洗脱的方式为梯度洗脱;
23.所述梯度洗脱的程序为:
24.0min:所述缓冲液f的体积百分含量为95%;所述缓冲液g的体积百分含量为5%;
25.0~20min:所述缓冲液f的体积百分含量由95%匀速减少到75%;所述缓冲液g的体积百分含量由5%匀速增加到25%;
26.优选的,所述去污剂包括lmng、digitonin和gdn中的一种或多种。
27.优选的,进行第三洗脱前,还包括:利用100kda截留分子量的浓缩管对所述第二纯化液进行浓缩。
28.优选的,所述人源线粒体粗提物的制备方法包括:破碎人源细胞,分离得所述人源线粒体粗提物。
29.本发明提供了一种人源atp合成酶的纯化方法,制备人源线粒体粗提物,溶膜,得线粒体总蛋白溶液;依次通过阳离子交换柱、阴离子交换柱及凝胶排阻色谱柱的手段分离纯化人源atp合成酶。本发明提供的纯化方法具有操作方法简单,实验成本低,时长短的特点,得到的人源atp合成酶蛋白纯度高,生理活性好,可用于后续的结构功能及药物发现研究。
附图说明
30.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍。
31.图1为sp阳离子交换柱图洗脱结果,横坐标为色谱柱洗脱体积,单位为ml;左纵坐标为280nm紫外吸收,单位为mau;右纵坐标为电导,单位为ms/cm;
32.图2为sp阳离子交换柱取样bn-page电泳结果;
33.图3为q阴离子交换柱洗脱结果,横坐标为色谱柱洗脱体积,单位为ml;左纵坐标为280nm紫外吸收,单位为mau;右纵坐标为电导,单位为ms/cm;
34.图4为q阳离子交换柱取样bn-page电泳结果;
35.图5为凝胶排阻色谱柱图洗脱结果,图中横坐标为色谱柱洗脱体积,单位为ml;纵坐标为280nm紫外吸收,单位为mau;
36.图6为样品sds-page图,左为marker,样品组分经过胶条质谱鉴定;
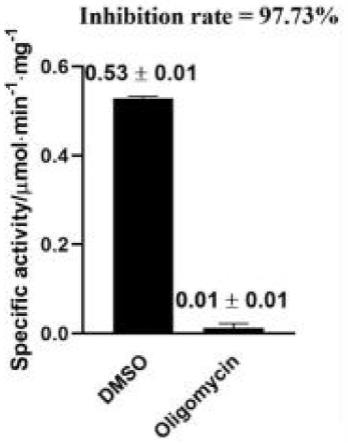
37.图7为atp水解活性测试。
具体实施方式
38.本发明提供了一种人源atp合成酶的纯化方法,包括如下步骤:
39.制备人源线粒体粗提物,溶膜,得线粒体总蛋白溶液;
40.利用阳离子交换柱对所述线粒体总蛋白溶液进行第一洗脱,收集洗脱液,得第一纯化液;
41.利用阴离子交换柱对所述第一纯化液进行第二洗脱,收集洗脱液,得第二纯化液;
42.利用凝胶排阻色谱柱对所述第二纯化液进行第三洗脱,得纯化的人源atp合成酶。
43.本发明对所述人源线粒体粗提物的来源并没有特殊限定,优选从人源细胞中提取得到,更优选包括:破碎人源细胞,分离得所述人源线粒体粗提物。本发明所述人源细胞优选为293f细胞;所述破碎的方式优选包括:将人源细胞与缓冲液a混合研磨。本发明优选使用玻璃组织匀浆器进行研磨。本发明所述缓冲液a优选包括250mm sucrose,20mm 3-(n-morpholino)propanesulfonic acid(mops)和1mm ethyleneglycoltetraacetic acid(egta);所述缓冲液a的ph优选为7.4。本发明所述缓冲液a提供合适的渗透压,将人源细胞与缓冲液a混合研磨后使人源细胞裂解。
44.在本发明中,所述研磨的次数优选为两次。本发明优选将所述人源细胞与缓冲液a混合进行第一次研磨,分离得第一上清液和第一沉淀。本发明进行所述第一次研磨时,所述缓冲液a的体积优选为人源细胞(沉淀)体积的5倍。本发明所述分离的方式优选为离心,所述离心的转速优选为1000g,所述离心的的时间优选为10min。
45.得所述第一沉淀后,本发明优选将所述第一沉淀用缓冲液a重悬所述第一沉淀,进行第二次研磨,分离得第二上清液和第二沉淀。本发明进行所述第二次研磨时,所述缓冲液a的体积优选为第一沉淀体积的5倍。本发明所述分离的方式优选为离心,所述离心的转速优选为1000g,所述离心的时间优选为10min。
46.得所述第二上清液后,本发明优选将所述第一上清液和第二上清液混合,分离得第三上清。本发明优选对所述第三上清液进行第三离心,所述第三离心的转速优选为10000g,时间优选为10min。本发明所述第三离心能够获得线粒体沉淀。
47.本发明直接以陈述的人源细胞系为实验材料,直接从人源细胞分别得到线粒体,实验样本大,成本低,并且操作简单。
48.本发明优选利用缓冲液b重悬所述人源线粒体粗提物,溶膜,得线粒体总蛋白溶液。本发明所述缓冲液b优选包括20mm 2-(n-吗啡啉)乙磺酸(mes)和50mm nacl;所述缓冲液b的ph优选为6.0。本发明所述溶膜的方式优选包括加入去污剂;所述去污剂优选包括lmng、digitonin和gdn中的一种或多种,进一步优选为lmng或digitonin;本发明所述溶膜的时间优选为30min~120min,进一步优选为50min~100min,更优选为60~80min。本发明所述人源线粒体粗提物与所述去污剂的体积比优选为临界胶束浓度的2~4倍;所述临界胶束浓度指所述去污剂形成胶束的最小浓度,由去污剂本身的性质决定,因此不作具体限定。本发明在具体实施过程中,以去污剂lmng为例进行说明,但是不能仅将其理解为本发明的全部保护范围。
49.溶膜后,本发明优选进行离心处理,得所述线粒体总蛋白溶液。本发明所述离心的转速优选为40000g;所述离心的时间优选为40min。本发明所述去污剂能够去除不溶物质。
50.得线粒体总蛋白溶液后,本发明利用阳离子交换柱对所述线粒体总蛋白溶液进行第一洗脱,收集洗脱液,得第一纯化液。本发明所述阳离子交换柱优选为强阳离子交换柱,进一步优选为sp阳离子交换柱或s阳离子交换柱;所述阳离子交换柱的体积优选为5ml。
51.本发明优选利用缓冲液b平衡所述阳离子交换柱,过滤所述线粒体总蛋白溶液的不溶物后进行所述第一洗脱。本发明所述缓冲液b的组成已在前记载,在此不作赘述。本发明优选使用缓冲液b平衡5个柱体积,即当所述阳离子交换柱的体积为5ml时,使用25ml缓冲液b平衡。本发明所述过滤优选使用0.45μm孔径的滤膜。
52.本发明优选利用缓冲液c和缓冲液d进行所述第一洗脱。本发明所述缓冲液c优选包括20mm 2-(n-吗啡啉)乙磺酸和0.004%w/v去污剂;所述缓冲液c的ph为6.0;本发明所述所述缓冲液d包括20mm 2-(n-吗啡啉)乙磺酸、1mnacl和0.004%w/v去污剂;所述缓冲液d的ph为6.0。本发明所述缓冲液c和缓冲液d中使用的去污剂优选与所述溶膜时使用的去污剂一致。所述第一洗脱的流速优选为1ml/min;所述第一洗脱的方式优选为梯度洗脱。本发明在第一洗脱过程中,优选保持阳离子交换柱的温度为4℃,以保持蛋白活性。本发明所述收集80~180mm nacl位置的洗脱液,得到所述第一纯化液。本发明所述第一纯化液中含有atp合成酶。本发明所述梯度洗脱的程序优选为:
53.0min:所述缓冲液c的体积百分含量为95%;所述缓冲液d的体积百分含量为5%;
54.0~40min:所述缓冲液c的体积百分含量由95%匀速减少到75%;所述缓冲液d的体积百分含量由5%匀速增加到25%。
55.得所述第一纯化液后,本发明优选利用阴离子交换柱对所述第一纯化液进行第二洗脱,收集洗脱液,得第二纯化液。本发明所述阴离子交换柱优选为强阴离子交换柱,进一步优选为q阴离子交换柱;所述阴离子交换柱的体积优选为1ml。
56.本发明优选利用缓冲液e平衡所述阴离子交换柱后进行所述第二洗脱。本发明优选使用缓冲液e平衡5个柱体积,即当所述阴离子交换柱的体积为1ml时,使用5ml缓冲液e平衡。本发明所述缓冲液e优选包括20mm mops、50mm nacl和0.004%w/v去污剂;所述缓冲液e的ph优选为7.4。本发明所述缓冲液e中使用的去污剂优选与所述溶膜时使用的去污剂一致。
57.本发明优选利用缓冲液f和缓冲液g进行所述第二洗脱。本发明所述缓冲液f优选包括20mm mops和0.004%w/v去污剂;所述缓冲液f的ph优选为7.4;本发明所述缓冲液g优选包括20mm mops、1m nacl和0.004%w/v去污剂;所述缓冲液d的ph优选为7.4。本发明所述缓冲液f和缓冲液g中使用的去污剂优选与所述溶膜时使用的去污剂一致。所述第二洗脱的流速优选为1ml/min;所述第二洗脱的方式优选为梯度洗脱。本发明在第二洗脱过程中,优选保持阴离子交换柱的温度为4℃。本发明所述收集110~190mm nacl位置的洗脱液,得到所述第二纯化液。本发明所述第二纯化液中含有atp合成酶。本发明所述梯度洗脱的程序为:
58.0min:所述缓冲液f的体积百分含量为95%;所述缓冲液g的体积百分含量为5%;
59.0~20min:所述缓冲液f的体积百分含量由95%匀速减少到75%;所述缓冲液g的体积百分含量由5%匀速增加到25%;
60.得所述第二纯化液后,本发明优选利用100kda截留分子量的浓缩管将第二纯化液的体积浓缩至≤500μl,得浓缩液。
61.得浓缩液后,本发明利用凝胶排阻色谱柱对所述浓缩液进行第三洗脱,得纯化的人源atp合成酶。本发明所述凝胶排阻色谱柱的体积优选为25ml。本发明优选利用缓冲液h平衡所述凝胶排阻色谱柱后进行所述第三洗脱。本发明优选使用缓冲液h平衡1个柱体积,
即当所述凝胶排阻色谱柱的体积为25ml时,使用25ml缓冲液h平衡。本发明所述缓冲液h优选包括20mm mops、100mm nacl和0.004%w/v去污剂;所述缓冲液h的ph优选为7.4。本发明所述缓冲液h中使用的去污剂优选与所述溶膜时使用的去污剂一致。
62.本发明采用阴-阳离子交换与分子筛连用的方式,具有纯度高、步骤简单、耗时短、安全性好以及易规范化操作等优点,与目前常用的密度梯度离心法相比,对实验设备要求低,大大减少了时间成本及资金成本,并且本发明纯化方法所用阴阳离子填料及分子筛填料简单易得,纯化流程易放大,可以进一步推进大规模分离纯化。
63.为了进一步说明本发明,下面结合附图和实施例对本发明提供的技术方案进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
64.实施例1
65.实施过程中使用的缓冲液:
66.缓冲液a:250mm sucrose,20mm 3-(n-morpholino)propanesulfonic acid(mops),1mm ethyleneglycoltetraacetic acid(egta),ph7.4;
67.缓冲液b:20mm mes,50mm nacl,ph 6.0;
68.缓冲液c:20mm mes,0.004%w/v lmng,ph 6.0;
69.缓冲液d:20mm mes,1m nacl,0.004%w/v lmng,ph 6.0;
70.缓冲液e:20mm mops,50mm nacl,0.004%w/v lmng,ph 7.4
71.缓冲液f:20mm mops,0.004%w/v lmng,ph 7.4;
72.缓冲液g:20mm mops,1m nacl,0.004%w/v lmng,ph 7.4;
73.缓冲液h:20mm mops,100mm nacl,0.004%w/v lmng,ph7.4。
74.(一)线粒体准备
75.培养3l人源悬浮细胞系hek293f至细胞密度为3.5
×
106个/ml,收获细胞体积为40ml。加入100ml的磷酸缓冲液(pbs)重悬细胞,1000g离心10min,收集细胞沉淀。用pbs重复洗涤一次。加入缓冲液a重悬细胞,总体积为200ml;用玻璃组织匀浆器上下研磨20次(一上一下记为一次),1000g离心10min,收集上清液;加入缓冲液a重悬沉淀,总体积200ml,再次研磨10次,1000g离心10min,收集上清液。将两次所得上清液合并,再次1000g离心10min,收集上清。上清液10000g离心10min,收集线粒体沉淀。
76.(二)线粒体总蛋白溶液准备
77.利用缓冲液b重悬线粒体沉淀至40ml,置于冰上并缓慢摇晃溶膜30min。溶膜结束后,40000g离心40min,收取上清液。使用0.45μm滤膜过滤上清液。
78.(三)阳离子交换柱
79.将hitrap sp强阳离子交换柱(5ml,ge healthcare)用缓冲液b平衡5个柱体积。将准备好的蛋白样品上样至离子柱,设置流速为1ml/min。以下操作使用akta pure系统。设置流速为1ml/min,使用缓冲液c和d设置50~250mm nacl线性梯度进行洗脱,具体洗脱程序为:0min:所述缓冲液c的体积百分含量为95%;所述缓冲液d的体积百分含量为5%;(0~40min:所述缓冲液c的体积百分含量由95%匀速减少到75%;所述缓冲液d的体积百分含量由5%匀速增加到25%。洗脱结果如图1所示,图1中横坐标为强阳离子交换柱洗脱体积,单位为ml。左纵坐标为280nm紫外吸收,单位为mau。右纵坐标为电导,单位为ms/cm,图中f为收集管编号,f1为第1收集管,f27为第27收集管。根据图1可以看出,前20ml洗脱体积出于平衡
阶段,电导在20ml处开始上升,最终收集的洗脱体积为40ml。
80.对sp阳离子交换柱取样进行blue native page(参考invitrogen native page 4-16%bis-tris gel说明书)电泳,检测如图2所示,根据图2并结合电导(cond)可以看出,atp合成酶在电导80-180ms/cm位置洗脱。
81.收集含有atp合成酶的组分。加入缓冲液f将蛋白溶液nacl浓度稀释为约50mmnacl,用1m tris调节ph至7.2-7.4。含有atp合成酶的蛋白溶液使用0.45μm滤膜过滤。
82.(四)阴离子交换柱
83.将hitrap q强阴离子交换柱(1ml,ge healthcare)用缓冲液e平衡5个柱体积。将准备好的蛋白样品上样至离子柱,设置流速为1ml/min。以下操作使用pure系统。设置流速为1ml/min,使用缓冲液f和g设置50-250mm nacl线性梯度进行洗脱,具体洗脱程序为:0min:所述缓冲液f的体积百分含量为95%;所述缓冲液g的体积百分含量为5%;0~20min:所述缓冲液f的体积百分含量由95%匀速减少到75%;所述缓冲液g的体积百分含量由5%匀速增加到25%。洗脱结果如图3所示,图3中横坐标为强阴离子交换柱洗脱体积,单位为ml。左纵坐标为280nm紫外吸收,单位为mau。右纵坐标为电导,单位为ms/cm,图中f为收集管编号,f1为第1收集管,f13为第13收集管。根据图3可以看出,前10ml洗脱体积出于平衡阶段,电导在10ml处开始上升,最终收集的洗脱体积为20ml。
84.对阴离子交换柱取样进行blue native page(invitrogen native page 4-16%bis-tris gel)电泳,检测如图4所示,根据图4可以看出,atp合成酶在110-190mmnacl位置洗脱。
85.收集含有atp合成酶的组分。将蛋白溶液用100kda截留分子量的浓缩管浓缩至500μl以下,得蛋白浓缩液。
86.(五)凝胶过滤色谱柱
87.用缓冲液h平衡superose6 increase10/300(ge healthcare)凝胶排阻色谱柱,将蛋白浓缩液上样并洗脱,结果如图5所示,图中横坐标为色谱柱洗脱体积,单位为ml。纵坐标为280nm紫外吸收,单位为mau。根据图5可以看出,atp合成酶在13.5ml位置出现特征峰;
88.收集特征峰位置样品进行sds-page电泳,检测如图6所示,根据图6可以看出,凝胶排阻色谱柱洗脱样品无明显杂条带,经质谱鉴定atp合成酶亚基组成完整。
89.实施例2
90.根据参考文献(guo,hui et al.structure of mycobacterial atp synthase bound to the tuberculosis drug bedaquiline.nature vol.589,7840(2021):143-147.),对实施例1特征峰位置样品进行atp水解活性测试,结果如图7所示,其中图7左侧为对照组,对照组为dmso;右侧为实验组,实验组为寡霉素(oligomycin),对照组dmso的体积与实验组寡霉素的体积一致。根据图7记载可以看出,本发明得到的纯化样品可被寡霉素(oligomycin)抑制,说明本发明得到的纯化样品中含有较高的atp合成酶。
91.本发明提供的纯化方法具有操作方法简单,实验成本低,时长短的特点,得到的人源atp合成酶蛋白纯度高,寡霉素抑制率高,生理活性好,可用于后续的结构功能及药物发现研究。
92.尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例,而不是全部实施例,人们还可以根据本实施例在不经创造性前提下获得其他实施例,这些
实施例都属于本发明保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1