一种微量游离DNA甲基化建库方法、试剂盒及测序方法与流程
一种微量游离dna甲基化建库方法、试剂盒及测序方法
技术领域
1.本发明涉及基因检测技术领域,具体而言,涉及一种微量游离dna甲基化建库方法、试剂盒及测序方法。
背景技术:
2.dna甲基化5mc是甲基转移酶介导的甲基供体s-腺苷酰甲硫氨酸(sam)分子中的甲基基团转移到dna胞嘧啶上的过程。在真核生物中,dna甲基化主要发生在cpg二核苷酸胞嘧啶的5碳位上,形成5-甲基胞嘧啶(5mc)。dna甲基化修饰对基因的表达调控起重要作用,是癌症发生发展、治疗预后重要的分析标志物。
3.cfdna是游离于细胞外的dna(cell-free dna,cfdna),片段长度通常在150bp左右,早在1984年由mandel和m
é
tais发现。肿瘤患者分离的游离dna由于大部分来源于肿瘤细胞,携带着各病变器官或组织的所有遗传变异信息,因此也称为ctdna。cfdna甲基化检测,特别是ctdna甲基化应用于肿瘤早期筛查、患者预后风险评估的标志物筛查鉴定,已经成为全球各国科学家和企业届争分夺秒竞相争夺解决的技术难题之一。其中最为核心的是游离dna的含量极低,片段长度短小,个体差异大。健康个体每毫升血液含有0-100ng,平均30ng/ml,即便是晚期肿瘤患者,由于大量肿瘤细胞的凋亡和坏死,其含量平均也仅为180ng/ml。产业方面,国内外各基因检测公司,为了快速建立起具有独立知识产权的检测技术,经常融资数亿美元用于研发,如美国的grail公司融资超10亿美元布局血浆游离dna在乳腺癌早期筛查的研发。国内也有多家单位已完成数亿的融资,着手开发相关的检测技术。
4.目前应用于游离dna组学筛选的检测技术主要是全基因组甲基化测序,例如grail公司投资的,2020年发表的基于细胞外游离dna甲基化特征,用于多肿瘤检测和定位的研究,主要就是通过全基因组甲基化测序鉴定关键的甲基化标志物。然而,由于游离dna的片段特征仅为140bp左右,即使测序90g的数据,平均深度也仅为15x,绝大部分的检测位点测序深度低于5x,单样本检测费用近万元,成本极高,一般单位难于进行大样本测序。此外,加拿大玛嘉烈公主癌症中心近年研发的cfmedip技术,可以对低至1ng的游离dna进行甲基化测序,其基本原理是通过甲基化特异性抗体捕获含甲基化修饰胞嘧啶碱基的dna片段,通过对捕获的片段测序,确定基因组不同片段的甲基化相对含量,该技术无法检测碱基的精准甲基化修饰水平,且具有捕获偏好性。
技术实现要素:
5.本发明所要解决的技术问题是提供一种微量游离dna甲基化建库方法、试剂盒及测序方法。
6.本发明解决上述技术问题的技术方案如下:
7.本发明提供一种微量游离dna甲基化建库方法,包括以下步骤:
8.1)提取并分离游离dna样本;
9.2)对所述游离dna样本进行双末端酶切修剪,得到游离dna片段;
10.3)在所述游离dna片段的5’端连接含有甲基化和生物素修饰的接头序列;
11.4)采用磁珠对连接接头序列的所述游离dna片段进行捕获和纯化,并得到产物;
12.5)采用亚硫酸盐转化液对所述步骤4)得到的产物进行亚硫酸盐转化;
13.6)对亚硫酸盐转化后的产物进行二链延伸;
14.7)对二链延伸的产物进行pcr扩增,得到dna甲基化文库。
15.进一步,所述步骤2)中,双末端酶切修剪采用的酶为mspi、alui、bstni、hpych4v、haeiii、hpych4iii、apeki、banii、sphi、bsssi、bglii、bamhi、kpni中的一种。
16.进一步,所述步骤3)中的所述接头序列为ad1,所述步骤3)还包括在所述游离dna片段的3’端连接ad2的步骤;所述步骤5)中,进行亚硫酸盐转化后,ad2结合在磁珠上并与所述游离dna片段分离,连接有ad1的所述游离dna片段转移至所述亚硫酸盐转化液中。
17.进一步,所述接头序列为基于illumina平台设计的序列;
18.其中,ad1的核苷酸序列如seq id no 1.所示,ad2的核苷酸序列如seq id no 2.所示;或,ad1的核苷酸序列如seq id no 3.所示,ad2的核苷酸序列如seq id no 4.所示。
19.进一步,所述接头序列为基于mgi平台设计的序列;
20.其中,ad1的核苷酸序列如seq id no 5.所示,ad2的核苷酸序列如seq id no 6.所示;或,中ad1的核苷酸序列如seq id no 7.所示,ad2的核苷酸序列如seq id no 8.所示。
21.进一步,所述步骤1)中提取并分离的游离dna样本量为0.3-2ng。
22.进一步,所述步骤6)中,进行二链延伸的试剂包括dna聚合酶,所述dna聚合酶为t4 dna polymerase、bst dna polymerase、klenow frgment(3
’‑5’
exo-)、pcr polymerase、hemo klentaq(neb)、amplitaq dna polymerase、stoffel fragment(life)和neb的(exo
–
)dna聚合酶、dna聚合酶中的一种。
23.进一步,所述步骤7)中,对二链延伸的产物进行pcr扩增后,还包括质量检测的步骤;所述质量检测的步骤检测扩增后的产物片段长度,当产物片段长度为150bp-300bp,则得到的产物为dna甲基化文库。
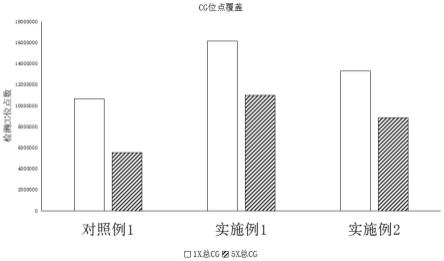
24.本发明提供一种微量游离dna甲基化建库试剂盒,用于如上述的微量游离dna甲基化建库方法;
25.包括游离dna样本的提取和分离试剂、酶切试剂、含有甲基化和生物素修饰的接头序列、接头序列连接试剂、亚硫酸盐转化试剂、二链延伸试剂以及pcr扩增试剂。
26.本发明提供一种游离dna测序方法,先采用如上述的微量游离dna甲基化建库方法进行建库,再进行测序。
27.本发明的有益效果在于:
28.1)本发明的微量游离dna甲基化建库方法,通过对酶切后的游离dna片段的5’端进行接头,使片段化的游离dna中的几乎所有ccgg位点都可被测序检测,并且增加了位点两侧的cg甲基化信息。
29.2)本发明的微量游离dna甲基化建库方法,对游离dna片段接头后,进行亚硫酸盐转化并对转化产物随机延伸,减少了dna的投入量,保证每一条测序序列均包含至少一个cg位点的甲基化信息,减少纯化丢失,使起始量dna尽可能多的被检测到,达到低起始量的目的,从而实现在进行微量检测的同时,还能够实现精确检测。
30.3)本发明的微量游离dna甲基化建库方法,游离dna样本量可低至小于1ng,与现有技术相比大幅度降低了样本量的需求。
31.4)本发明的微量游离dna甲基化建库方法,能够显著增加游离dna的cg检测位点数量,对于cg位点的覆盖效果显著。
32.5)本发明的游离dna测序方法,相比于全基因组cfdna甲基化测序,测序深度显著增加,测序数据量更低,成本更低。
附图说明
33.图1为本发明的微量游离dna甲基化建库方法中,实施例1的步骤1中,检测cfdna提取效果的色谱图;
34.图2为本发明的游离dna测序方法中,实施例1、实施例2以及对比例1的测序结果中,1x及5x cg位点覆盖的数量和占比图。
具体实施方式
35.以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
36.dna由于降解的原因,其片段长度显著减少,常规的简化基因组甲基化测序很难富集到足够的cg位点并进行甲基化测序。特别像cfdna等样本,其自身片段长度通常在150bp左右,完全包含在简化基因组甲基化技术检测的片段范围(40bp-300bp)之内。而含有ccgg位点的片段通过简化基因组甲基化技术处理,有效片段被进一步酶切至更短而丢失,反而使片段中不含ccgg位点的片段却保留下来并被测序,浪费有效数据,减少了cg位点检测数量。
37.本发明的游离dna甲基化建库方法能够针对微量的游离dna进行建库的原理在于,本发明通过对游离dna酶切产物5’端连接接头,理论上经过酶切的几乎所有ccgg位点都可被检测,且增加位点两侧的cg甲基化信息。相比于全基因组游离dna甲基化测序,本发明采用先酶切、再接头的片段化处理方式,能够使对于游离dna甲基化的测序深度显著增加,测序数据量更低,成本更低。
38.本发明的微量游离dna甲基化建库方法,包括以下步骤:
39.1)提取并分离游离dna样本;
40.本发明的建库方法主要针对血液中的游离dna,即cfdna,或肿瘤患者血液中循环肿瘤细胞游离dna,即ctdna;也可针对正常组织或细胞基因组dna,即gdna。
41.本发明的游离dna样本可来源于外周血、脑脊液,唾液、支气管灌洗液、胸腹腔积液、胃液、胆汁、粪便、尿液等。
42.优选的,本发明的cfdna样本来源于血浆。
43.本发明的样本量可低于1ng并低至单细胞水平,优选的,本发明的样本量在0.3ng~2ng之间。
44.其中,dna的投入量可低于1ng或单细胞水平。样本dna的提取可根据不同样本性质选用已有商业化提取试剂盒或实验室自建技术进行提取。
45.优选的,最后提取游离dna样品的洗脱只能用超纯水或eb缓冲液,不能使用te或成
分不明的试剂盒自带洗脱液洗脱,样品洗脱体积不超过10ul,防止缓冲液对游离dna样品造成干扰。
46.优选的,对于cfdna甲基化检测,为了避免胞内dna的甲基化信号干扰,样本提取后可进行毛细管电泳检测cfdna片段的分布特征,提取效果好的cfdna产物片段应主要分布在140bp左右,可在280bp附近有小量dna存在,1000bp以上应不存在基因组dna片段。
47.2)采用mspi酶对游离dna样本进行双末端修剪,得到游离dna片段。
48.dna片段在mspi酶的作用下进行双末端修剪,优选的,反应总体系为10-50ul,构建5’端凸出cg碱基的片段结构,底物在37℃反应20min-16h。
49.酶切体系具体为:
[0050][0051]
3)在游离dna片段的5’端连接含有甲基化修饰的接头序列。
[0052]
在步骤2)得到的游离dna片段中,分别加入连接buffer a、甲基化和生物素共同修饰的接头及dna连接酶,16-30℃反应20min-6h。
[0053]
接头序列中含有生物素修饰,能够使后续步骤中的磁珠更好的对游离dna片段进行捕获。
[0054]
反应体系具体为:
[0055][0056]
buffer a的成分具体为
[0057][0058]
其中,接头序列包括ad1和ad2,两个接头序列均与游离dna片段连接;
[0059]
接头序列为基于illumina平台设计的序列或基于mgi平台设计的序列;其中,基于illumina平台设计的序列中,ad1的核苷酸序列如seq id no 1.所示,ad2的核苷酸序列如seq id no 2.所示。
[0060]
基于mgi平台设计的序列中ad1的核苷酸序列如seq id no 5.所示,ad2的核苷酸序列如seq id no 6.所示。
[0061]
优选的,基于illumina平台设计的序列还可以有以下替代方案:ad1的核苷酸序列如seq id no 3.所示,ad2的核苷酸序列如seq id no 4.所示;
[0062]
基于mgi平台设计的序列还可以有以下替代方案:ad1的核苷酸序列如seq id no 7.所示,ad2的核苷酸序列如seq id no 8.所示。
[0063]
基于illumina平台设计的序列,其中,seq id no 1.有两种表示方式,第一种表示方式为,5
’‑
ggagttcagacgtgtgctcttccgatct-3’,其中,划横线的碱基为甲基化的碱基;第二种表示方式为,
[0064]5’‑
ggagttmcagamcgtgtgmctmcttmcmcgatmct-3’,m为甲基,表示位于其之后的碱基甲基化。
[0065]
seq id no 2.:5
’‑
cgagatcggaagagcacacgtctgaactcc-bio-3’。
[0066]
可代替为含有index的接头序列中,seq id no 3.有两种表示方式,第一种表示方式为,5
’‑
ggagttcagacgtgtgctcttccgatctnnnnnnnn-3’,其中,划横线的碱基为甲基化的碱基,n为任意碱基;第二种表示方式为,5
’‑
ggagttmcagamcgtgtgmctmcttmcmcgatmctnnnnnnnn-3’,m为甲基,表示位于其之后的碱基甲基化,n为任意碱基。
[0067]
seq id no 4.:5
’‑
cgnnnnnnnnagatcggaagagcacacgtctgaactcc-bio-3’。
[0068]
基于mgi测序接头设计的序列中,seq id no 5.有两种表示方式,第一种表示方式为,5
’‑
ttgtcttcctaaggaacgacatggctacgatccgactt-3’,其中,划横线的碱基为甲基化的碱基;第二种表示方式为,5
’‑
ttgtcttcctaaggaamcgamcatggmctamcgatmcmcgamctt-3’,m为甲基,表示位于其之后的碱基甲基化。
[0069]
seq id no 6.:
[0070]5’‑
cgaagtcggatcgtagccatgtcgttccttaggaagacaa-bio-3’。
[0071]
可代替的接头序列中,seq id no 7.有两种表示方式,第一种表示方式为,5
’‑
gaacgacatggctacgatccgactt-3’,其中,划横线的碱基为甲基化的碱基;第二种表示方式为,5
’‑
gaamcgamcatggmctamcgatmcmcgamctt-3’,m为甲基,表示位于其之后的碱基甲基化。
[0072]
seq id no 8.:5
’‑
cgaagtcggatcgtagccatgtcgttc-bio-3’。
[0073]
4)采用磁珠对连接接头的所述游离dna片段进行捕获和纯化,并得到磁珠-游离dna捕获混合液。
[0074]
本步骤采用链霉素标记的磁珠捕获连接甲基化和生物素共同修饰的接头的游离dna连接产物,捕获产物重悬至20ul超纯水中。具体的操作步骤如下:
[0075]
首先,取20-40ul的链霉素偶联磁珠,用50ul的漂洗液漂洗三次,磁珠重悬于15ul的eb缓冲液中。
[0076]
其次,将游离dna接头连接产物加入15ul的磁珠中,室温孵育10-30min,磁力架捕获游离dna连接片段;
[0077]
最后,用50ml的eb漂洗磁珠2次,磁力架去干净上清后,将磁珠重悬到20ul的超纯水或0.1m的氢氧化钠中。
[0078]
需要说明的是,如果采用超纯水进行洗脱,本步骤最后得到的是磁珠-游离dna混合液。如用氢氧化钠等强碱替换超纯水溶解磁珠,则将洗脱上清进行回收,去磁珠,得到不含有磁珠的上清液,将该上清液直接进行下一步亚硫酸盐转化。
[0079]
5)对步骤4)得到的产物进行亚硫酸盐转化。
[0080]
采用商业化试剂盒对游离dna进行亚硫酸盐转化。
[0081]
如果步骤4)中采用的是超纯水进行的洗脱,那么在按照试剂盒说明书实验前,首先在20ul的磁珠捕获液中加入试剂盒推荐体积的亚硫酸盐反应液,98℃变性1-10min,将游离dna从磁珠变性下来,16000rpm离心5-10min或磁力架静置,将上清转移到一个新的pcr管中。
[0082]
如果用0.1m的氢氧化钠洗脱,洗脱液放到磁力架上,吸取上清20ul到pcr管中,加入试剂盒推荐体积的亚硫酸盐反应液后,直接按照试剂盒说明书进行亚硫酸盐转化。反应后,将c-t转化后的游离dna洗脱至26ul的超纯水或eb中。
[0083]
经过该步骤的亚硫酸盐转化后,接头序列ad2结合在磁珠上,连接接头序列ad1的cfdna片段变性到亚硫酸盐转化液中。
[0084]
6)对亚硫酸盐转化后的游离dna片段进行二链延伸。
[0085]
在亚硫酸盐转化后dna的pcr管中,分别加入以下试剂,对亚硫酸盐转化产物进行二链延伸。
[0086][0087]
采用primer1序列设计引物特征,使用含有以下特征的引物进行二链延伸。
[0088]
基于illumina测序平台设计的引物序列如seq id no 9.所示,其中,n为随机碱基:
[0089]
oligo 1:5
’‑
tacacgacgctcttccgatctnnnnnn-3’(seq id no 9.)。
[0090]
基于mgi测序平台设计的引物序列如seq id no 10.所示,s为index序列,n为随机碱基:
[0091]
oligo 1:5
’‑
tgtgagccaaggagttgssssssssssttgtcttcctaagaccgcttggcctccgacttnnnnnn-3’(seq id no 10.)。
[0092]
具体的操作步骤为:
[0093]
将上述反应液混匀后,置于pcr仪,95℃加热45s,反应后快速转移冰上静置5min,加入dna聚合酶klenow(3
′→5′
exo-)100u,混匀后,置于pcr仪上4℃孵育5min,孵育后按照0.1-1℃/s升温速率升温至37℃,随后37℃孵育1.5h,反应后用1倍磁珠进行纯化,洗脱至20ul的eb缓冲液中。
[0094]
该反应dna聚合酶包括但不限于t4 dna polymerase、bst dna polymerase、klenow frgment(3
’‑5’
exo-)、pcr polymerase、hemo klentaq(neb)、amplitaq dna polymerase、stoffel fragment(life)和neb的(exo
–
)dna聚合酶、dna聚合酶等。
[0095]
本发明的建库方法中,经过步骤2)的酶切后,通过步骤3)的接头单端连接,再经过步骤4)、5)完成亚硫酸盐转化后,进行本步骤的对亚硫酸盐转化产物随机延伸,这样的方法减少了dna的投入量,保证每一条测序序列均包含至少一个cg位点的甲基化信息,减少纯化
丢失,使起始量dna尽可能多的被检测到,达到低起始量的目的。
[0096]
7)对二链延伸的产物进行pcr扩增。
[0097]
本步骤采用的引物为含有以下特征的引物:
[0098]
对于illumina测序平台,pcr扩增的上游引物(primer1)和下游引物(primer2)的序列特征分别如seq id no 11.和seq id no 12.所示,其中,n为随机碱基:
[0099]
primer 1:5
′‑
aatgatacggcgaccaccgagatctacacnnnnnnnnacactctttccctacacgacgctcttccgatct-3’(seq id no 11.);
[0100]
primer 2:5
′‑
caagcagaagacggcatacgagatnnnnnnnngtgactggagttcagacgtgtgc-3’(seq id no 12.)。
[0101]
对于mgi测序平台,pcr扩增的上游引物(primer1)和下游引物(primer2)的序列特征分别如seq id no 13.和seq id no 14.所示,其中,n为随机碱基,其primer 2的5端进行磷酸化修饰:
[0102]
primer 1:5
’‑
tgtgagccaaggagttg-3’(seq id no 13.);
[0103]
primer 2:5phos-gaacgacatggctacga-3’(seq id no 14.)。
[0104]
本步骤的pcr扩增体系为:
[0105][0106][0107]
pcr扩增条件如下:
[0108]
98℃,1min;98℃,30s,50-68℃,30s,72℃,30s,10-18个循环;72℃,5mim。
[0109]
pcr扩增纯化后,洗脱至20ul的缓冲液中。
[0110]
pcr扩增后,还包括质量检测的步骤;所述质量检测的步骤检测扩增后的产物片段长度,当产物片段长度为150bp-300bp,则得到的产物为游离dna甲基化文库。如果产物片段长度不在上述范围内,则建库失败,需要重新进行建库。
[0111]
本发明还提供一种dna测序方法,在采用上述步骤建立dna甲基化文库后,对得到的文库进行测序。
[0112]
具体步骤为,对采用本发明的建库方法建库得到的dna甲基化文库,通过qpcr定量样本dna的莫尔浓度,毛细管电泳或2100生物分析仪检测文库片段大小后,合格片段为150bp-270bp之间,根据测序数据量取相应摩尔的文库,在测序仪上测序。
[0113]
测序后,对下机数据分别进行去接头、低质量值reads过滤和统计、甲基化转换率统计、比对及比对率统计、基因组覆盖度统计、重复率统计,并计算每一位点甲基化水平,得到cg位点覆盖数。
[0114]
以下通过具体的实施例对本发明的方法进行说明:
[0115]
实施例1
[0116]
本实施例提供一个具体的采用本发明的方法进行建库和测序的步骤。具体过程为:
[0117]
1)样品收集及cfdna提取:在含edta的采血管或cfdna采血管中收集全血2-5ml,颠倒混匀,1600g离心后,立即将上层血浆部分,进一步16000g 4℃离心10min,去除残余细胞及碎片,分离上清血浆至新的离心管中,-80度保存。
[0118]
收集血浆运用商业化cfdna提取试剂盒进行提取。采用的试剂盒为dsp blood mini kit(qiagen),qubit hs定量提取dna的浓度,并采用安捷伦2100检测cfdna提取效果,判断是否存在基因组污染。当不存在基因组污染时,进行下一步。
[0119]
图1为检测cfdna提取效果的色谱图,通过图1可以看出,本实施例提取的cfdna不存在基因组污染。
[0120]
2)mspi酶切cfdna片段:取1-2ng的无基因组污染的游离dna,按照以下体系进行ccgg位点酶切:mspi酶50u,10x buffer 1ul,补水至总体积10ul,37℃反应2小时。
[0121]
3)cfdna末端连接接头序列:在上述cfdna酶切体系中依次加入1um的接头序列ad1和ad2退火产物1ul,3.5ul的buffer a和200u的dna连接酶,混匀后,23℃,孵育2小时。
[0122]
本实施例采用的接头序列为基于illumina平台设计的序列中,ad1的核苷酸序列,即如seq id no 1.和如seq id no 2.所示的序列。
[0123]
4)接头连接产物捕获:取40ul的链霉素偶联磁珠,用50ul的漂洗液漂洗三次,磁珠重悬于15ul的eb缓冲液中;其次,将cfdna接头连接产物加入15ul的磁珠中,室温孵育30min,磁力架捕获cfdna连接片段;用100ml的eb漂洗磁珠2次,磁力架去干净上清后,将磁珠重悬到20ul的超纯水中
[0124]
5)接头ad2去除及亚硫酸盐处理:应用zymo research公司的ez dna methylation-gold
tm
kit亚硫酸盐转化试剂盒进行亚硫酸盐处理。
[0125]
处理流程如下:将20ul的磁珠-cfdna捕获混合液中加入到130ul的亚硫酸盐反应液中,98℃变性8min,16000rpm离心10min,接头ad2仍结合在磁珠上,而cfdna变性到亚硫酸盐转化液中,将上清转移到一个新的pcr管内,在pcr仪上继续按照一下程序进行反应:98℃,2min;64℃,2.5小时,反应后按照试剂盒说明书进行纯化,洗脱至26ul的超纯水中。
[0126]
6)对亚硫酸盐转化后的cfdna片段进行二链延伸:在纯化的26ul转化后dna中,分别加入5ul的10x反应缓冲液,2ul的10mm的dntp,和5ul的20mm的oligo 1,补水至总体积50ul,将上述反应液混匀后,置于pcr仪,95℃加热45s,反应后快速转移冰上静置5min,加入dna聚合酶klenow(3
′→5′
exo-)100u,混匀后,置于pcr仪上4℃孵育5min,孵育后按照1℃/s升温速率升温至37℃,随后37℃孵育1.5h,反应后用1倍dna纯化磁珠进行纯化,洗脱至20ul的eb缓冲液中。
[0127]
7)pcr扩增
[0128]
在pcr管中,按照以下体系配制pcr反应液
[0129][0130]
pcr反应条件为:95℃预变性1min;95℃,30s;58℃,30s;72℃,45s进行12个循环扩增,72℃延伸5min,16℃保存;pcr产物扩增后,用1倍体积的ampure xp beads纯化,洗脱至20ul的eb洗脱液中。
[0131]
文库构建后,分别运用安捷伦2100进行片段大小检测及qpcr浓度定量检测,合格后,运用hiseq进行测序。
[0132]
对本实施例的文库分别进行两侧测序,测序深度分别为1x和5x。
[0133]
实施例2
[0134]
采用与实施例1相同的步骤和试剂对gdna进行建库和测序。对本实施例的文库分别进行两侧测序,测序深度分别为1x和5x。
[0135]
对比例1
[0136]
采用常规的简化甲基化方法对gdna进行建库和测序。对本对比例的文库分别进行两侧测序,测序深度分别为1x和5x。
[0137]
分别考察实施例1和2以及对比例1的测序结果中,1x和5x测序深度下,cg位点覆盖的数量和占比,结果如图2所示。
[0138]
通过图2的结果可以看出,实施例1和2的1x cg位点覆盖分别为16.2m和13.3m,占基因组总cg位点的23%和24%;而常规简化基因组甲基化1xcg位点覆盖仅为10m左右,占基因组cg位点约16%。5x cg位点覆盖分别为10.97m和8.83m;而常规简化基因组甲基化1xcg位点覆盖仅为5.56m左右。
[0139]
因此,本发明的建库方法及测序方法,适用于微量cfdna或gdna的检测,对于cg位点的覆盖效果显著。
[0140]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1