一种滇龙胆中性多糖及其制备方法与应用
1.本发明属于天然产物提取技术领域,具体涉及一种滇龙胆中性多糖及其制备方法与应用。
背景技术:
2.多糖(polysaccharide)是一类天然大分子物质,由单糖之间脱水缩合,通过糖苷键连接而成的聚合物。与核酸、蛋白质一样,是生命活动的基础物质之一。多糖具有多种生理功能,广泛参与细胞识别、胚胎发育、细胞分化、生长、代谢、病毒感染、免疫应答等生命活动,具有抗肿瘤、抗凝血、抗氧化、抗突变、抗病毒、降血糖、抗溃疡、降血脂等生物活性,在药品及保健食品领域有很多的研究与应用。
3.滇龙胆(gentiana rigescens franch.ex hemsl.)为龙胆科龙胆属多年生宿根小草本植物,是我国常用中药之一,具有清热燥湿,泻肝胆火等功效。目前,对滇龙胆的有效成分的研究较多,但针对滇龙胆中多糖的研究未见报道,本发明旨在提供一种自滇龙胆中提取的具有显著的抗炎及抗氧化活性的多糖。
技术实现要素:
4.本发明的第一目的是提供一种滇龙胆中性多糖;本发明的第二目的是提供一种滇龙胆中性多糖的制备方法,本发明的第三目的是提供一种滇龙胆中性多糖的应用。
5.本发明的第一目的是这样实现的,所述滇龙胆中性多糖由岩藻糖:鼠李糖:阿拉伯糖:半乳糖:葡萄糖:木糖:甘露糖:葡萄糖醛酸摩尔比为3.544:6.973:4.635:31.364:37.149:1.962:12.190:2.182,分子量为3074 da,所述滇龙胆中性多糖的主要衍生物为3,4,6-me3-man(p)、3,4,6-me4-glc (p)以及2,3,6-me3-gal(p)组成,主要连接方式为
→
1)
‑ꢀ
man(p)-(2
→
、
→
1)
‑ꢀ
gal(p)-(2
→
以及
→
1)
‑ꢀ
gal(p)-(4
→
。
6.本发明的第二目的是这样实现的,所述滇龙胆多糖的制备方法按以下步骤实现:(1)将干燥的滇龙胆药材,粉碎为粗粉,过3号筛(50目筛),石油醚索氏回流脱脂2-3次后,以80-85℃水提,每次提取2.5-3 h,提取次数为2-3次,合并提取液,减压浓缩得到浸膏;(2)将步骤1得到的浸膏以无水乙醇醇沉,离心,弃去上清液,将沉淀用蒸馏水重溶,以sevege法除蛋白,离心,弃去沉淀,共5-6次,直至离心再无沉淀,减压浓缩,冷冻干燥,得到滇龙胆粗多糖;(3)将步骤2得到的滇龙胆粗多糖用蒸馏水溶解,离心过滤后,采用deae sepharose fast flow阴离子交换层析纯化,以去离子水洗脱,收集洗脱液,经透析、浓缩后得到中性多糖;本发明的第三目的是这样实现的,所述滇龙胆中性多糖的应用为作为活性成分或者是药用载体在制备抗炎症药物中的应用。
7.本发明的有益效果为:
(1)本发明提供了一种从滇龙胆中提取的中性多糖,所述中性多糖具有较高的抗炎及抗氧化活性,其抑制tnf-α及il-6分泌的最低浓度为25μg/ml,所述中性多糖在浓度为6.4 mg/ml时就已经具有了78.5%的vc的dpph自由基清除能力。另外,本发明提供的中性多糖毒副作用小,因此,在制备低毒高效的抗炎及抗氧化药品开发中具有广泛的应用前景,有利于滇龙胆资源的进一步开发利用。
8.(2)本发明制备中性多糖的方法不仅操作简单成本低廉,而且更加高效实用,同时更好的保护了多糖的生物活性,值得推广应用。
附图说明
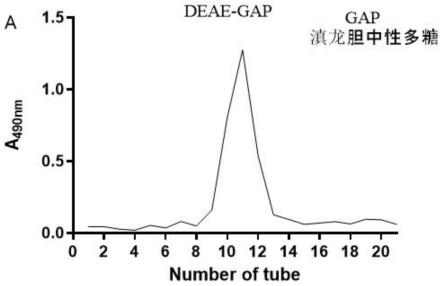
9.图1为本发明滇龙胆中性多糖gap的葡萄糖标准曲线(y=0.0138x-0.0265 (r2=0.9946));图2为本发明滇龙胆中性多糖gap的纤维素阴离子交换柱分离谱图;图3为本发明滇龙胆中性多糖gap的气质联用色谱仪数据图;图4为本发明滇龙胆中性多糖gap 的hpgpc色谱图;图5为本发明滇龙胆中性多糖gap的总离子流图;图6为本发明滇龙胆中性多糖gap 的红外光谱图;图7为本发明滇龙胆中性多糖gap细胞活力数据图;图8为本发明滇龙胆中性多糖gap抑制脂多糖诱导的raw 264.7巨噬细胞产生促炎细胞因子il-6的作用示意图;图9为本发明滇龙胆中性多糖gap抑制脂多糖诱导的raw 264.7巨噬细胞产生促炎细胞因子tnf-α的作用示意图;图10为本发明滇龙胆中性多糖gap及维生素c对dpph自由基的清除作用的曲线图;图11为白云参中性多糖(cjp-c)以及白云参酸性多糖(cjp)dpph自由基清除能力曲线图。
具体实施方式
10.下面结合附图和实施例对本发明作进一步的说明,但不以任何方式对本发明加以限制,基于本发明教导所作的任何变更或改进,均属于本发明的保护范围。
11.本发明提供了一种滇龙胆中性多糖,所述中性多糖由岩藻糖:鼠李糖:阿拉伯糖:半乳糖:葡萄糖:木糖:甘露糖:葡萄糖醛酸摩尔比为3.544:6.973:4.635:31.364:37.149:1.962:12.190:2.182,分子量为3074 da,所述滇龙胆中性多糖的主要衍生物为3,4,6-me3-man(p)、3,4,6-me4-glc (p)以及2,3,6-me3-gal(p)组成,主要连接方式为
→
1)
‑ꢀ
man(p)-(2
→
、
→
1)
‑ꢀ
gal(p)-(2
→
以及
→
1)
‑ꢀ
gal(p)-(4
→
。
12.本发明滇龙胆中性多糖的制备方法按以下步骤实现:(1)将干燥的滇龙胆药材粉碎为粗粉,过3号筛(50目筛),经石油醚索氏回流脱脂2-3次后,以80-85℃水提,每次提取2.5-3 h,提取次数为2-3次,合并提取液,减压浓缩得到浸膏;(2)将步骤1得到的浸膏以无水乙醇醇沉,离心,弃去上清液,将沉淀用蒸馏水重溶,以sevege法除蛋白,离心,弃去沉淀,共5-6次,直至离心再无沉淀,减压浓缩,冷冻干燥,
得到滇龙胆粗多糖;(3)将步骤2得到的滇龙胆粗多糖用蒸馏水溶解,离心过滤后,采用deae sepharose fast flow阴离子交换层析纯化,以去离子水洗脱,收集洗脱液,经透析、浓缩后得到目标滇龙胆中性多糖。
13.步骤1中,石油醚索氏回流脱脂的温度为60-90 ℃,石油醚的加入量为滇龙胆药材重量的6-8倍(v:m=6-8)。
14.步骤1中,热水煮提的温度为80-85℃。
15.步骤2中,无水乙醇浓度为100%,无水乙醇的加入量为所述浸膏的2-3倍(v:m=2-3)。
16.步骤3中,透析时的截流分子量为3400 da。
17.本发明滇龙胆中性多糖的应用为作为活性成分或者是药用载体在制备抗炎症药物中的应用。
18.所述应用将所述滇龙胆多糖加入药学上可接受的辅料制备成片剂、硬胶囊、软胶囊、散剂、丸剂、颗粒剂。
19.实施例1将1kg滇龙胆药材粉碎后,60℃下用6 l石油醚索氏回流脱脂3次。将脱脂后的滇龙胆药材以10 l 80 ℃的热水进行煮提,每次提取3小时,共提取2次,合并提取液,减压浓缩。将浓缩后的浸膏以2倍体积 无水乙醇醇沉,离心,弃去上清液,将沉淀用蒸馏水重溶,以sevege法除蛋白,离心,弃去沉淀,共5次,直至离心再无沉淀。最后将除蛋白后的多糖减压浓缩,冷冻干燥,得到80.92g滇龙胆粗多糖。
20.利用deae sepharose fast flow阴离子交换层析纯化所述滇龙胆粗多糖(分离图谱如图2所示),取所述滇龙胆粗多糖10.08 g溶于少量蒸馏水中,配制成30 mg/ml的溶液,4000rpm离心12min,上清液用0.45 μm滤头过滤,上样;用去离子水洗脱,洗脱5倍柱体积,流速为1 ml/min,接洗脱液,减压浓缩至1/15的体积;浓缩后的洗脱液装于分子量3400 da的透析袋中,放于蒸馏水中透析24 h,每5h换一次水;得到的洗脱液冷冻干燥得0.86 g滇龙胆中性多糖,得率或收率为8.53%。
21.实施例2将1kg滇龙胆药材粉碎后,75℃下用7 l石油醚索氏回流脱脂3次。将脱脂后的滇龙胆药材以12 l 85℃的热水进行煮提,每次提取2.5小时,共提取3次,合并提取液,减压浓缩。将浓缩后的浸膏以2.5倍体积 无水乙醇醇沉,离心,弃去上清液,将沉淀用蒸馏水重溶,以sevege法除蛋白,离心,弃去沉淀,共6次,直至离心再无沉淀。最后将除蛋白后的多糖减压浓缩,冷冻干燥,得到100.12 g滇龙胆粗多糖。
22.利用deae sepharose fast flow阴离子交换层析纯化所述滇龙胆粗多糖,取所述滇龙胆粗多糖10.15 g溶于少量蒸馏水中,配制成30 mg/ml的溶液,5000rpm离心8min,上清液用0.45 μm滤头过滤,上样;用去离子水洗脱,洗脱5倍柱体积,流速为0.9ml/min,接洗脱液,减压浓缩至1/16的体积;浓缩后的洗脱液装于分子量3400 da的透析袋中,放于蒸馏水中透析24 h,每3 h换一次水;得到的洗脱液冷冻干燥得1 .08 g滇龙胆中性多糖,得率或收率为10.64%。
23.实施例3
将1kg滇龙胆药材粉碎后,85℃下用8 l石油醚索氏回流脱脂3次。将脱脂后的滇龙胆药材以12l 85℃的热水进行煮提,每次提取3小时,共提取3次,合并提取液,减压浓缩。将浓缩后的浸膏以3倍体积无水乙醇醇沉,离心,弃去上清液,将沉淀用蒸馏水重溶,以sevege法除蛋白,离心,弃去沉淀,共6次,直至离心再无沉淀。最后将除蛋白后的多糖减压浓缩,冷冻干燥,得到121.86g滇龙胆粗多糖。
24.利用deae sepharose fast flow阴离子交换层析纯化所述滇龙胆粗多糖:取所述滇龙胆粗提物10.09 g溶于少量蒸馏水中,配制成35 mg/ml的溶液,于4500rpm下离心9min,上清液用0.45 μm滤头过滤,上样;用去离子水洗脱,洗脱6倍柱体积,流速为0.8 ml/min,接洗脱液,减压浓缩至1/10的体积;浓缩后的洗脱液装于分子量3400 da的透析袋中,放于蒸馏水中透析28 h,每7 h换一次水;得到的洗脱液冷冻干燥得1.28g滇龙胆中性粗多糖,得率为12.69%。
25.将本发明制备得到的滇龙胆中性多糖命名为gap,以下以实施例1为例,对gap进行结构及活性等分析检测。
26.试验例1 滇龙胆中性多糖gap中多糖的含量测定测定方法:苯酚-硫酸法(1)6 %苯酚溶液的配制:准确称量苯酚固体15g,加入250ml蒸馏水于60℃水浴下充分溶解,用棕色磨口瓶避光保存备用。
27.(2)0.1mg/ml gap溶液的制备:准确称量滇龙胆中性多糖10mg,加蒸馏水定容至100ml容量瓶中,配制成0.1mg/ml的gap溶液,待测。
28.(3)0.1 mg/ml 葡萄糖标准溶液的配制:准确称量葡萄糖标准品10 mg,加蒸馏水定容至100 ml容量瓶中,配制成0.1 mg/ml的葡萄糖标准溶液,待用。
29.(4)葡萄糖标准曲线的绘制:用移液枪分别量取葡萄糖标准溶液0 ml、0.2 ml、0.4 ml、0.6 ml、0.8 ml、1.0 ml于12ml玻璃试管中,加蒸馏水补至1ml,每个浓度3个重复。接下来向每个试管中加入0.5ml 6 %的苯酚溶液,继续缓慢加入2.5 ml浓硫酸,快速震荡试管,自然冷却待其颜色变化稳定后,于490 nm波长下测量其吸光度。以葡萄糖标准品质量为横坐标,吸光度a为纵坐标,绘制标准曲线(图1)。
30.(5)多糖样品的含量测定:取1 ml 0.1 mg/ml gap溶液,加入0.5 ml 6 %苯酚溶液,2.5 ml浓硫酸溶液,于490 nm波长下测量其吸光度。根据标准曲线计算出滇龙胆中性多糖的含量。
31.表1 苯酚-硫酸法测定实施例2制备的gap的吸光度结果如表1所示,可知本实施例制得的多糖含量为92.31 %。
32.试验例2 滇龙胆中性多糖gap单糖组成鉴定鉴定方法:分别准确称取实施例1中制备得到的gap及各单糖标准品(岩藻糖 (fuc)、鼠李糖(rha)、阿拉伯糖(ara)、半乳糖(gal)、葡萄糖(glc)、木糖(xyl)、甘露糖
(man)、果糖(fru)、核糖(rib)、半乳糖醛酸(galua)、葡萄糖醛酸(glcua)、甘露糖醛酸(manua)、古罗糖醛酸(gul-ua)各2 mg至各血清瓶中,其中单糖标准品进行单标及混标的衍生化反应。称取10 mg盐酸羟胺及1 mg内标肌醇,然后加入2 ml吡啶,于90 ℃条件下反应30 min,待其自然冷却后,再加入2 ml醋酸酐,拧紧瓶盖于90 ℃条件下反应30 min,待反应结束自然冷却后加入2 ml蒸馏水终止反应。向衍生化反应后的样品中加入2 ml二氯甲烷,充分震荡后让其静置分层,吸取下层溶液转移至25 ml蒸馏烧瓶中,再用1 ml二氯甲烷重复萃取1次,将两次萃取的溶液减压浓缩至干以除去多余水分,过0.22 μm有机滤膜,再进行gc-ms联用色谱检测。
33.结果分析:将各保留时间与单糖标准品比对后(图3),可确定其所含的单糖组成及摩尔百分比为:岩藻糖:鼠李糖:阿拉伯糖:半乳糖:葡萄糖:木糖:甘露糖:葡萄糖醛酸=3.544:6.973:4.635:31.364:37.149:1.962:12.190:2.182。
34.试验例3 滇龙胆中性多糖gap分子量测定称取样品gap以及不同分子量的右旋糖酐标准品各5 mg,分别加入0.05 m nacl溶液配制为5 mg/ml的供试品及标准品溶液,使用0.22 μm的微孔滤膜过滤,转至进样小瓶中冷藏备用。采用hpgpc法,使用高效液相色谱仪,示差检测器,聚合物基质水溶性sec(gfc)色谱柱ohpak sb-803 hq、ohpak sb-804 hq、ohpak sb-805 hq(8
×
300 mm)串联柱检测,流动相为0.05m nacl溶液,流速0.6ml/min,柱温40
ꢀº
c,进样量30μl。使用高效凝胶渗透色谱串联柱来进行供试品及标准品检测,采用waters empower软件对结果进行分析,以相应色谱峰的保留时间为横坐标,以标准品相对分子质量mp的对数值为纵坐标进行线性回归,建立标准曲线,将供试品保留时间代入标准曲线,计算可得供试品相对分子质量为3074 da(图4)。
35.试验例4 滇龙胆中性多糖gap甲基化分析称取10 mg gap,加入1 ml一级水溶解,加入1 ml 100 mg/ml碳二亚胺,反应2h,继续加入1ml 2m的咪唑,将样品平均分为两份,分别加入1 ml 30 mg/ml 的nabh4和1ml 30mg/ml 的nabd4,反应3 h,后加入100μl冰醋酸终止反应。透析样品48h,透析完成后冷冻干燥样品。对冻干样品进行甲基化处理,于冻干样品中加入500μl dmso溶解,加入1mg naoh,孵育30min,继续加入50μl碘甲烷溶液反应1h,后加入1ml水和2ml二氯甲烷,涡旋混匀,离心,弃水相。重复水洗3次,吸取下层二氯甲烷相并蒸干。继续加入100ml 2m tfa,121℃反应90 min后于30 ℃下蒸干,之后加入50μl 2m 氨水,50μl 1m nabd4,混匀,室温下反应2.5 h后,加入20μl乙酸终止反应,氮气吹干,250μl甲醇洗两次,氮气吹干。加入乙酸酐250μl,涡旋混匀,100℃反应2.5 h,继续加入1ml水静置10 min,再加入500μl二氯甲烷,涡旋混匀,离心,弃水相。重复水洗3次,反应完成后取下层二氯甲烷相,待测。色谱系统采用的是agilent气象色谱系统(agilent 7890a;agilent technologies,usa),根据化合物的性质,进样量为1μl,分流比10:1,载气为高纯氦气;柱温箱的初始温度为140℃保持2.0min,以3℃/min程序升温至230℃,保持3 min。质谱系统采用的是美国aiglent公司的四极杆质谱检测系统(agilent 5977b;agilent technologies,usa),配有电子轰击离子源(ei)和masshunter工作站。采用电子轰击离子源(ei),分析物在全扫描(scan)模式下进行检测,质量扫描范围(m/z): 30-600。将样品溶液按照上述色谱、质谱条件进样检测,获得多糖甲基化后特征性碎片,根据已有的数据库进行对比,进而确认其键合方式,gap主要衍生物为3,
4,6-me3-man(p)、3,4,6-me4-glc(p)以及2,3,6-me3-gal(p)组成,主要连接方式为
→
1)-man(p)-(2
→
、
→
1)-gal(p)-(2
→
以及
→
1)-gal(p)-(4
→
。结果如表2及图5所示:表2gap衍生物及键合方式试验例5 滇龙胆中性多糖gap红外分析实验方法:称取 2mg gap与 kbr 充分混合研磨后压成薄片于 ftir 仪中进行检测,检测波长为 4000 nm-400 nm。
36.测定结果如图6所示,3403.58 cm-1
处的信号峰是-oh伸缩振动产生的;2890.55cm
‑1、2931.60cm-1
处的信号峰是c-h伸缩振动产生的;1638.76cm-1
、处的信号峰是c=o的非对称产生的;1418.89cm-1
处的吸收峰是c-o伸缩振动产生的;1043.65cm-1
、1019.87cm-1
处的信号峰(1000cm-1
~1200cm-1
)这个区域是两种co伸缩振动产生的,分别是c-o-h和糖环的c-o-c;938.11cm-1处的吸收峰是β型糖苷键的特征峰;817.92cm-1
处的信号吸收峰是吡喃糖α-端基差向异构的c-h变角振动。
37.试验例6gap细胞活力检测检测方法:raw264.7细胞于dmem(含10%fbs,1%青霉素-链霉素)培养于37℃含5%co2的培养箱。实验分组如下:正常对照组(nc)、0.1μg/mllps处理组(阳性对照组)及不同浓度的滇龙胆中性多糖均一组分gap(10-1000μg/ml)处理组,每个分组设置6个复孔。将细胞接种于96孔培养板中,接种浓度为1
×
105个/ml,每孔100μl,培养24小时后,吸弃原培养基,正常组加入dmem培养基,其余各组加入对应浓度的药物,培养24h后,加入10μlcck8试剂,于培养箱中培养1.5小时后,450nm下用酶标仪检测吸光度,实验重复3次。细胞活力计算公式如下:细胞存活率=(as
−
ab)/(ac
−
ab)
×
100%(as:实验孔、ac:对照孔、ab:空白孔)。
38.测定结果如图7所示,与nc组细胞对比gap(10-1000μg/ml)处理组细胞活力均上升,且gap剂量在10μg/ml-250μg/ml最为显著(p<0.0001)。
39.结果分析:上述试验结果表明gap可以促进巨噬细胞raw264.7的增殖,且无毒副作用。
40.试验例7gap抗炎活性测定实验方法:将raw264.7接种于96孔板中,接种浓度为1
×
105个/ml,每孔100ul。培养24小时后,设置正常对照组(nc)、1μm/l地塞米松处理组(阳性对照组)、0.1μg/mllps(模型组)及不同浓度gap(25-100μg/ml)每个浓度设置4个复孔,正常组和模型组给予完全培养基,每个药物组每孔给予100ul的含药培养基培养24h。24小时后,吸弃细胞上清,正常组加入dmem培养基,其余各组均加入0.1μg/mllps,每孔0.1ml,培养12小时后,收集细胞上清液,进行elisa法检测细胞上清中的tnf-α及il-6的水平。
41.检测结果如图8和图9所示,其中,数据为三个独立实验的平均值
±
sd。单用脂多糖可显著诱导raw264.7巨噬细胞产生tnf-α和il-6(p《0.0001、p《0.001、p<0.05)。相反,gap治疗以剂量依赖的方式抑制这些lps刺激的促炎细胞因子的分泌(图7-8,p《0.0001、p《0.001、p<0.05)。
42.注:与nc组(正常组)相比,“####”表示p《0.0001;与mc组(模型对照组)相比,“*”表示p《0.05,“**”表示p<0.01,“***”表示p《0.001,“****”表示p《0.0001。
43.实验结果:1、由图8可知,模型组为经过脂多糖刺激的巨噬细胞产生大量的促炎细胞因子il-6,经过浓度为1μm/l的阳性药物地塞米松干预后有显著降低il-6分泌,gap在浓度为25μg/ml时已经初步对il-6的分泌水平有抑制作用,在浓度为100μg/ml的时候,可以极显著抑制由lps刺激引起的il-6的分泌水平。
44.2、由图9可知,模型组为经过脂多糖刺激的巨噬细胞产生大量的促炎细胞因子tnf-α,经过浓度为1μm/l的阳性药物地塞米松干预后有显著降低tnf-α分泌,gap在浓度为
25μg/ml时已经初步对tnf-α的分泌水平有抑制作用,在浓度为100μg/ml的时候,可以极显著抑制由lps刺激引起的tnf-α的分泌水平。
45.结果分析:本发明采用了脂多糖激活的巨噬细胞raw264.7炎症反应模型,通过对促炎细胞因子指标il-6、tnf-α的检测,评价其体外抗炎活性,实验结果表明滇龙胆中性多糖能够显著降低促炎细胞因子水平,其具有较好的抗炎活性。
46.试验例8gap抗氧化活性测定实验方法:dpph自由基清除能力测定准确称取2mgdpph溶解于10ml无水乙醇后定容于50ml容量瓶中(0.04mg/ml)于4℃下避光保存备用。将1ml浓度为0.2mg/ml、0.4mg/ml、0.6mg/ml、0.8mg/ml、1.6mg/ml、3.2mg/ml、6.4mg/ml的gap加入试管中,以等体积的蒸馏水为空白对照,分别向每个试管当中加入1mldpph溶液,避光反应30分钟,以维生素c作为阳性对照,于517nm下测定吸光度,试验结果如图10所示。自由基清除率计算公式如下:清除率(%)=[(ac-as)/ac]
×
100ac为空白吸光度,as为dpph溶液吸光度。
[0047]
实验结果:从图10可知,本发明滇龙胆中性多糖浓度在6.4mg/ml下的清除能力最大,且清除率达到78.49%,说明其具有极好的抗氧化能力。与现有技术白云参中性及酸性多糖dpph自由基清除能力相比,当浓度达到6.4mg/ml时,白云参中性及酸性多糖清除率才达到59.7%以及65.7%,本发明滇龙胆中性多糖的自由基清除能力更强,更加具有开发抗氧化药物的潜力。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1