一种DBCO修饰的聚乙二醇链接剂及其合成方法与流程
一种dbco修饰的聚乙二醇链接剂及其合成方法
技术领域
1.本发明涉及化学领域,尤其涉及一种dbco修饰的聚乙二醇链接剂及其合成方法。
背景技术:
2.随着大分子免疫类生物制药业、单克隆抗体偶联的药物定向传输技术、脂质体配方类和小型聚合物类纳米颗粒技术的发展,生物基因药物定向传输在抗癌类毒素药物开发中的应用逐步兴起并越来越繁荣。
3.聚乙二醇修饰的小分子药物可以改变化合物的理化性质,且大部分的前药是在达到特异性的作用靶点才被活化而发挥药效的,在很大程度上,聚乙二醇使的药物持续且稳定的作用于特异性的细胞或器官。
4.一般来讲,聚乙二醇修饰小分子药物分为两种:第一种是被动靶向性的药物释放系统,由于epr效应具有被动靶向性;第二种是靶向性的药物释放系统,由载体、活性化合物、靶分子组成,有时还需要连接臂将他们偶联起来。dbco修饰的小分子聚乙二醇链接剂,可以改变药物的理化性质,提高溶解度,达到缓释目的。
技术实现要素:
5.有鉴于此,本发明提出了一种dbco修饰的,能够改变药物的理化性质,提高溶解度,达到缓释目的的聚乙二醇链接剂及其制备方法。
6.本发明的技术方案是这样实现的:本发明提供了一种dbco修饰的聚乙二醇链接剂,分子通式为,
[0007][0008]
其中n为正整数;r为羟基、以及中的其中一种。
[0009]
另一方面,本发明还提供了一种dbco修饰的聚乙二醇链接剂的合成方法,当r为羟基时,合成步骤如下,
[0010][0011]
其中n为正整数。
[0012]
在以上技术方案的基础上,优选的,当r为及时,合成方法如下,
[0013]
将化合物10分别与及有机物发生反应得到化合物11至13;
[0014]
化合物11的分子式为:
[0015][0016]
化合物12的分子式为:
[0017][0018]
化合物13的分子式为:
[0019][0020]
其中n为正整数。
[0021]
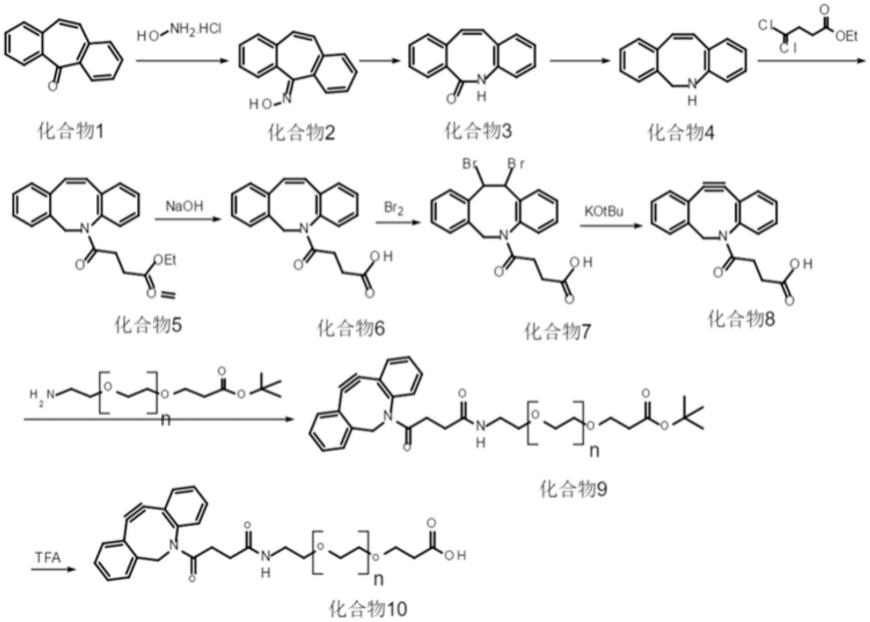
在以上技术方案的基础上,优选的,化合物2合成的具体方法为,将化合物1溶解于吡啶溶液中,然后滴加盐酸羟胺进行反应,待反应完全后,将反应液分离去除吡啶,倒入冰水,并加入浓盐酸和正己烷,过滤及干燥后得到化合物2;
[0022]
化合物3合成的具体方法为,将化合物2溶解于三氟乙酸内进行反应,待反应完全后,去除反应液中的溶剂得到化合物3;
[0023]
化合物4合成的具体方法为,将化合物3溶解于乙醚溶液中,然后滴加四氢锂铝进行反应,待反应完全后,反应液加水淬灭,过滤得到化合物4;
[0024]
化合物5合成的具体方法为,将化合物4溶解于二氯甲烷溶液中,然后向混合溶液中加入三乙胺混合均匀,再向内滴加丁二酸单乙酯酰氯进行反应,待反应完全后,反应液经萃取、去溶剂并分离提纯得到化合物5;
[0025]
化合物6合成的具体方法为,将化合物5溶解于四氢呋喃、甲醇和水的混合溶液中,然后滴加氢氧化钠进行反应,待反应完全后,将反应液分离去除甲醇和四氢呋喃,将反应液的ph值调整至酸性,再经过萃取、去溶剂和分离提纯得到化合物6;
[0026]
化合物7合成的具体方法为,将化合物6溶解于二氯甲烷溶液中,然后向內滴加溴素进行反应,待反应完全后,用硫代硫酸钠淬灭溴素,反应液再经过萃取、去溶剂和分离提纯得到化合物7;
[0027]
化合物8合成的具体方法为,将化合物7溶解于四氢呋喃溶液中,然后向內滴加含有叔丁醇钾的四氢呋喃溶液进行反应,待反应完全后,将反应液的ph值调整至酸性,反应液再经过萃取、去溶剂和分离提纯得到化合物8;
[0028]
化合物9合成的具体方法为,将化合物8溶解于二氯甲烷溶液中,再加入edci和dmap,最后加入15-氨基-4,7,10,13-四氧杂十五烷酸叔丁酯进行反应,待反应完全后,将反应液的ph值调整至酸性后,反应液再经过萃取、去溶剂和分离提纯得到化合物9;
[0029]
化合物9合成的具体方法为,将化合物9加入二氯甲烷和三氟乙酸的混合溶液中进行反应,待反应完全后,反应液经过分离提纯得到化合物10。
[0030]
进一步的,优选的,将及的其中一种与edci加入二氯甲烷溶液进行充分混合,然后向混合溶液内滴加化合物10进行反应,反应液再经过萃取、去溶剂和分离提纯得到化合物11至13中的其中一种。
[0031]
进一步的,优选的,化合物10与或者与或者与的质量比为(3-5):1。
[0032]
进一步的,优选的,化合物4至6均在零度环境下进行合成;化合物7在10至15度环境下进行合成;化合物8在-50至-40度环境下进行合成;其余化合物均在室温环境下进行合成。
[0033]
进一步的,优选的,化合物6、化合物8及化合物9的合成中,反应液在经过萃取及去溶剂前,将反应液的ph调至3至5。
[0034]
本发明的一种dbco修饰的聚乙二醇链接剂及其合成方法相对于现有技术具有以下有益效果:
[0035]
(1)本发明以5-二苯并环庚烯酮为原料,合成dbco,再与聚乙二醇链接起来,能够改变药物的理化性质,提高溶解度,达到缓释目的。
[0036]
(2)本发明的合成方法简单,反应条件温和,产生副反应较少,终产物收率高于90%,适合工业大规模生产,具有很好的经济效益和市场前景。
具体实施方式
[0037]
下面将结合本发明实施方式,对本发明实施方式中的技术方案进行清楚、完整地描述,显然,所描述的实施方式仅仅是本发明一部分实施方式,而不是全部的实施方式。基于本发明中的实施方式,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施方式,都属于本发明保护的范围。
[0038]
本发明中的化学式缩写:
[0039]
dbco:二苯并环辛炔
[0040]
kotbu:叔丁醇钾
[0041]
tfa:三氟乙酸
[0042]
dmap:4-二甲氨基吡啶
[0043]
edci:1-乙基-3(3-二甲基丙胺)碳二亚胺。
[0044]
在实施例二和三中1h nmr由varian 400在d2o中测得。
[0045]
实施例一:
[0046]
本发明提供了一种dbco修饰的聚乙二醇链接剂,分子通式为,
[0047][0048]
其中n为正整数;r为羟基、以及中的其中一种。
[0049]
实施例二:
[0050]
本发明还提供了一种实施例一中的dbco修饰的聚乙二醇链接剂的合成方法,当实施例一的分子通式中的r为羟基时,合成方法包括如下步骤,
[0051][0052]
其中n为正整数。
[0053]
其中,化合物1为5-二苯并环庚烯酮。
[0054]
化合物2的合成:在室温下,将化合物1(180g,0.873mol)溶于吡啶(630ml)溶液中,再将盐酸羟胺(92g,1.31mol)缓慢滴加到混合溶液中,加完120度回流过夜,tlc显示化合物1已全部反应完全;离心分离除去大部分吡啶,倒入1l冰水,加入100ml浓盐酸和200ml正己烷,滤出白色固体,充分干燥后称重有200g,计算收率为103.5%;产品无须进一步纯化直接以100%收率投往下一步。
[0055]
化合物3的合成:在室温下,将90ml三氟乙酸加入到化合物2(18.7g,84.6mmol)中,80度回流过夜后,tlc显示化合物2已全部反应完全;离心分离除去三氟乙酸,加入500ml水,过滤得到的固体依次用200ml dcm和100ml乙醇打浆,过滤得到灰色固体,充分干燥后称重有16g,计算收率为80%。
[0056]
化合物3的核磁共振:1h nmr(400mhz,dmso)δ11.34(s,1h),7.68
–
7.15(m,8h),6.95(d,j=0.7hz,2h).
[0057]
化合物4的合成:在零度环境下,将化合物3(14g,63.3mmol)溶于200ml乙醚溶液中,再将四氢锂铝(24g,0.63mol)缓慢滴加到混合溶液中,加完回流过夜,tlc显示化合物3已全部反应完全;反应液加水淬灭,过滤得到黄色固体,充分干燥后称重有12.60g,计算收率为96%。
[0058]
化合物4的核磁共振:1h nmr(400mhz,dmso)δ7.28
–
7.04(m,4h),6.97
–
6.69(m,2h),6.57
–
6.33(m,3h),6.28
–
6.14(m,2h),4.44(d,j=7.2hz,2h).
[0059]
化合物5的合成:在零度环境下,将化合物4(10g,48.24mmol)和三乙胺(5.85g,57.89mmol)溶于100ml二氯甲烷溶液中,再将丁二酸单乙酯酰氯(8.73g,53.06mmol)缓慢滴加到混合溶液中,加完室温反应1h,tlc显示反应已完全;向反应液加入200ml水,采用二氯
甲烷萃取,水洗3次后,干法快速过柱得到7.2g白色固体,收率89%。
[0060]
化合物6的合成:在零度环境下,将化合物5(10g,48.24mmol)溶于20ml四氢呋喃、20ml甲醇以及20ml水的混合溶液中,再将氢氧化钠(1.72g,42.93mmol)缓慢滴加混合溶液中,加完后室温反应2h,tlc显示反应已完全;反应液离心分离除去甲醇和四氢呋喃,将反应液的ph值调整为4后,向反应液中加入20ml水,再采用二氯甲烷萃取,水洗3次,干法快速过柱后,以100%收率投往下一步。
[0061]
化合物7的合成:在10度环境下,将化合物6(102.48g,0.333mol)溶于1.5l二氯甲烷溶液中,再将溴素(58.60g,0.367mol)缓慢滴加到混合溶液中,加完后室温反应2h,tlc显示反应已完全;向反应液加入200ml水,用硫代硫酸钠淬灭溴素,再采用二氯甲烷萃取,水洗3次,干法快速过柱,最后用600ml乙酸乙酯打浆得到129g白色固体,收率80.5%。
[0062]
化合物7的核磁共振:1h nmr(400mhz,cdcl3)δ7.73(d,j=7.7hz,1h),7.30
–
7.00(m,6h),6.90(d,j=7.5hz,1h),5.89(d,j=9.9hz,1h),5.82(d,j=14.9hz,1h),5.16(d,j=9.9hz,1h),4.21(d,j=14.9hz,1h),3.01
–
2.81(m,1h),2.73
–
2.44(m,3h).
[0063]
化合物8的合成:在-40度环境下,将化合物7(140g,299.6mmol)溶于四氢呋喃(1.2l)溶液中,再将含有叔丁醇钾(1.3l,1m)的四氢呋喃溶液缓慢滴加到混合溶液中,过程中分批滴加进行反应,并控制在3h内加完,tlc显示反应已完全;向反应液加入200ml水,调整反应液的ph值为4,采用二氯甲烷萃取,水洗3次,干法快速过柱,最后用300ml乙酸乙酯打浆得到88g浅黄色固体,收率96.7%。
[0064]
化合物8的核磁共振:1h nmr(400mhz,cdcl3)δ7.68(d,j=7.5hz,1h),7.50
–
7.18(m,7h),5.16(d,j=13.9hz,1h),3.70(d,j=13.8hz,1h),2.66(dddd,j=46.9,16.9,8.9,5.3hz,2h),2.36(ddd,j=16.9,6.3,5.3hz,1h),2.16
–
1.88(m,1h).
[0065]
化合物9的合成:在室温下,将化合物8(88.5g,0.29mol)溶于0.9l二氯甲烷中,在向其中依次加入edci(61.1g,0.319mol)和dmap(3.54g,29mmol),最后加入15-氨基-4,7,10,13-四氧杂十五烷酸叔丁酯(93g,0.29mol),室温反应过夜,tlc显示反应已完全;向反应液加入200ml水,调整反应液的ph值为4,采用二氯甲烷萃取,水洗3次,干法快速过柱得到156.14g黄色油状物,收率88.5%。
[0066]
化合物9的核磁共振:1h nmr(400mhz,cdcl3)δ7.61(d,j=7.3hz,1h),7.53
–
7.42(m,1h),7.41
–
7.07(m,6h),6.23(t,j=5.3hz,1h),5.08(d,j=13.8hz,1h),3.68
–
3.45(m,14h),3.39(dt,j=6.6,5.1hz,2h),3.28(t,j=5.3hz,2h),2.76(ddd,j=16.7,8.3,6.6hz,1h),2.46
–
2.33(m,3h),2.09(dt,j=15.3,6.2hz,1h),1.87(dt,j=16.8,6.2hz,1h),1.38(s,9h).
[0067]
化合物10的合成:在室温下,将化合物9(35g,57.57mmol)溶于150ml二氯甲烷和150ml三氟乙酸的混合溶液中,室温反应1h,tlc显示反应已完全;反应液干法快速过柱得到24.4g黄色油状物,收率77%。
[0068]
化合物10的结构式为,其中n为正整数。
[0069]
实施例三:
[0070]
在实施例二的基础上,当dbco修饰的聚乙二醇链接剂的分子通式中r为时,可通过以下步骤,合成获得化合物11。
[0071]
具体来说,合成步骤为,在室温下,将化合物n-羟基琥珀酰亚胺(8.58g,74.6mmol)和edci(19.40g,94.1mmol)溶于500ml二氯甲烷溶液中,再将化合物10(40g,72.4mol)缓慢滴加到混合溶液中,室温反应过夜,tlc显示反应已完全;向反应液加入200ml水,采用二氯甲烷萃取,水洗5次,干法快速过柱得到48.7g黄色油状物,收率100%。
[0072]
化合物11的结构式为,其中n为正整数。
[0073]
化合物11的核磁共振:1h nmr(400mhz,cdcl3)δ7.65(d,j=7.4hz,1h),7.56
–
7.43(m,1h),7.43
–
7.11(m,6h),6.30(s,1h),5.13(d,j=13.8hz,1h),3.89
–
3.20(m,19h),2.82(dt,j=23.3,10.6hz,7h),2.54
–
2.33(m,1h),2.14(dt,j=15.1,6.2hz,1h),1.92(dt,j=16.7,6.2hz,1h).
[0074]
实施例四:
[0075]
在实施例二的基础上,当dbco修饰的聚乙二醇链接剂的分子通式中r为时,可通过以下步骤,合成获得化合物12。
[0076]
具体来说,合成步骤为,在室温下,将化合物2,3,5,6-四氟苯酚(9.72g,81.6mmol)和edci(19.40g,94.1mmol)溶于500ml二氯甲烷溶液中,再将化合物10(40g,72.4mol)缓慢滴加到混合溶液中,室温反应过夜,tlc显示反应已完全;向反应液加入200ml水,采用二氯甲烷萃取,水洗5次,干法快速过柱得到53.4g黄色油状物,收率100%。
[0077]
化合物12的结构式为,其中n为正整数。
[0078]
实施例五:
[0079]
在实施例二的基础上,当dbco修饰的聚乙二醇链接剂的分子通式中r为时,可通过以下步骤,合成获得化合物13。
[0080]
具体来说,合成步骤为,在室温下,将化合物五氟苯酚(8.31g,76.8mmol)和edci(19.40g,94.1mmol)溶于500ml二氯甲烷溶液中,再将化合物10(40g,72.4mol)缓慢滴加到
混合溶液中,室温反应过夜,tlc显示反应已完全;向反应液加入200ml水,采用二氯甲烷萃取,水洗5次,干法快速过柱得到49.5g黄色油状物,收率100%。
[0081]
化合物13的结构式为,其中n为正整数。
[0082]
以上所述仅为本发明的较佳实施方式而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1