一种盐酸阿罗洛尔中间体的制备工艺的制作方法
1.本发明涉及药物化学制备领域,具体涉及一种盐酸阿罗洛尔中间体的制备方法。
背景技术:
2.盐酸阿罗洛尔由日本住友制药株式会社研发,1985年首次在日本上市。盐酸阿罗洛尔被称为第四代β受体阻滞剂,是β受体阻滞剂的一线药物。临床主要用于治疗轻至中度原发性高血压和心绞痛等。本品是一种选择性β1-肾上腺素受体拮抗剂,兼有微弱的α1-肾上腺受体阻滞作用,可在降压的同时抑制α-肾上腺素受体兴奋,降低交感神经的张力,降压效果更理想,更适用于青少年高血压的治疗。
3.盐酸阿罗洛尔的化学结构式如下:
[0004][0005]
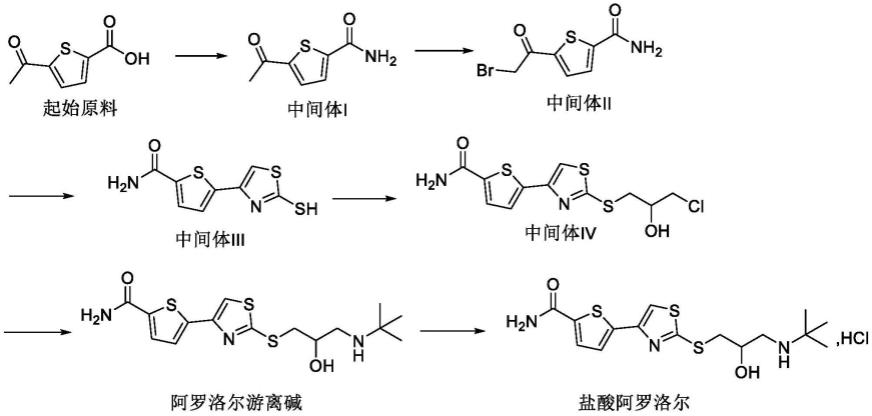
目前关于盐酸阿罗洛尔的制备方法的文献较多,其中主流合成路线如下:
[0006][0007]
其中,针对路线中中间体ⅱ的合成,文献“synthesis andβ-adrenergic blocking action of a new thiazolylthiopropanolamine derivative,journal of pharmaceutical sciences,1878,67(9),1334)”、文献“盐酸阿罗洛尔的合成,中国医药工业杂质.2011,42(9),641”、文献“盐酸阿罗洛尔合成工艺改进,药学进展,2013,37(3),137”及专利cn104447731a等报道的相关制备方法中,均采用溴素作为溴源,存在毒性大、需高温反应、杂质多、收率低、对设备腐蚀大等弊端。
[0008]
因此,开发一条反应条件温和、操作简单、毒性低、利于环保和劳动保护、适合工业化生产的中间体ⅱ制备工艺具有极为重要的意义。
技术实现要素:
[0009]
为了解决上述技术问题,本发明提供一种盐酸阿罗洛尔中间体的制备工艺,该工艺各步骤反应条件温和、反应步骤短、原子利用率高、操作简单可控、反应收率高、产品纯度高、不使用高毒性试剂,利用环保和劳动保护,适合于大规模工业化生产。
[0010]
本发明公开了一种盐酸阿罗洛尔中间体的制备工艺,包括如下步骤:
[0011][0012]
a)将起始原料在溶剂1中先与酰氯化试剂反应后,再与氨解试剂反应生成中间体ⅰ;
[0013]
b)将所得中间体ⅰ在溶剂2中与溴化试剂反应生成5-(2-溴乙酰)噻吩-2-甲酰胺。
[0014]
优选地,步骤a)中所述的溶剂1选自乙腈、二氯甲烷、四氢呋喃;
[0015]
更优选地,步骤a)中所述的溶剂1为二氯甲烷。
[0016]
优选地,步骤a)中所述的酰氯化试剂为草酰氯。
[0017]
优选地,步骤a)中所述的氨解试剂为氨水。
[0018]
优选地,步骤a)中所述的酰氯化反应温度为10~40℃;
[0019]
更优选地,步骤a)中所述的酰氯化反应温度为15~25℃。
[0020]
优选地,步骤a)中所述的氨解反应温度为-10~25℃;
[0021]
更优选地,步骤a)中所述的氨解反应温度为为-10~5℃。
[0022]
更进一步优选地,步骤a)中所述的氨解反应温度为-5~5℃。
[0023]
优选地,步骤a)中所述的起始原料与酰氯化试剂的摩尔比为1:1~1.5;
[0024]
更优选地,步骤a)中所述的起始原料与酰氯化试剂的摩尔比为1:1.3。
[0025]
优选地,步骤a)中所述的起始原料与氨解试剂的摩尔比为1:4~10;
[0026]
更优选地,步骤a)中所述的起始原料与氨解试剂的摩尔比为1:4.5。
[0027]
优选地,步骤b)中所述的溴化试剂选自三溴化吡啶鎓、四溴环酮、四丁基三溴化铵、三溴化苄基三甲基铵、溴合二氧六环、溴化溴代二甲基锍盐或5,5-二溴丙二酸亚异丙酯;
[0028]
更优选地,步骤b)中所述的溴化试剂选自三溴化吡啶鎓、四溴环酮、四丁基三溴化铵;
[0029]
优选地,步骤b)中所述的反应温度为30~50℃;
[0030]
更优选地,步骤b)中所述的反应温度为35~45℃。
[0031]
优选地,步骤b)中所述的反应溶剂2为冰醋酸;
[0032]
优选地,步骤b)中所述的中间体ⅰ与反应溶剂2的质量体积比为1:10~15;
[0033]
更优选地,步骤b)中所述的中间体ⅰ与反应溶剂2的质量体积比为1:15。
[0034]
优选地,步骤b)中所述中间体ⅰ与溴化试剂的摩尔比为1:0.9~1;
[0035]
更优选地,步骤b)中所述中间体ⅰ与溴化试剂的摩尔比为1:0.96。
[0036]
与现有技术相比,本发明的技术方案取得以下有益效果:
[0037]
(1)现有技术中,由中间体ⅰ制备5-(2-溴乙酰)噻吩-2-甲酰胺的过程,多以溴素作为溴源或在对甲苯磺酸催化下用nbs上溴。以溴素作为溴源,存在毒性大、需高温反应、杂质多、收率低、对设备腐蚀大等弊端;而用nbs上溴,需用大量对甲苯磺酸催化,反应的副产物较多(对甲苯磺酸的甲基上可能上溴),且可能引入潜在的遗传毒性杂质。与之相比,本发明的反应条件温和,无需使用催化剂,不涉及高毒性试剂和基因毒性杂质问题,副反应少,后处理方法简单,反应试剂对设备不具有腐蚀性,产物纯度高,纯度可达到90%以上,符合绿色经济化学的概念,降低了生产与环保成本,适合于工业化大规模生产。
[0038]
(2)本发明的合成步骤少,仅涉及两步化学反应,原子利用率高,在克服现有技术反应试剂毒性大、副反应多、存在潜在的遗传毒性杂质等缺陷的同时,所得中间体及产物的收率与现有技术相当或更高,中间体ⅰ收率为95.15%-96.29%,终产物5-(2-溴乙酰)噻吩-2-甲酰胺收率为91.03%-96.15%,整条路线的总收率可达到85%以上。
具体实施方式
[0039]
下面通过实施例来进一步说明本发明。应该正确理解的是:本发明的实施例中的方法仅仅是用于说明本发明而给出的,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属于本发明要求保护的范围。
[0040]
本发明中所涉及的核磁共振仪(1h nmr)为bruker avance-400,核磁共振(1h nmr)位移(δ)以百万分之一(ppm)的单位给出,内标为四甲基硅烷(tms),化学位移是以10-6
(ppm)作为单位给出。
[0041]
质谱仪器的型号为tsq quantum ultra液质联用仪。
[0042]
实施例1:5-(2-溴乙酰)噻吩-2-甲酰胺的制备
[0043]
(a)中间体ⅰ的制备:
[0044]
将11.5kg5-乙酰-噻吩-2-羧酸悬浮于152.4kg二氯甲烷中,加入230gn,n-二甲基甲酰胺,氮气置换后搅拌降温至0℃,缓慢加入11.15kg草酰氯。加毕,缓慢升温至15~25℃搅拌。反应完全后,将反应液减压浓缩至干。残余物加入152.4kg二氯甲烷,搅拌溶解完全后,备用。向反应釜中加入23l浓氨水,降温至-5~5℃,缓慢加入上述中间态——酰氯的二氯甲烷溶液。加毕,离心,滤饼用水淋洗。减压干燥得到10.98kg中间体ⅰ,收率96.06%,hplc纯度99.72%。
[0045]1h-nmr(dmso-d6,δtms0):2.51ppm(3h,t,j=1.6),7.71ppm(1h,s),7.78ppm(1h,d,j=4.0),8.20ppm(1h,s),7.92ppm(1h,d,j=4.0)
[0046]
(b)5-(2-溴乙酰)噻吩-2-甲酰胺的制备:
[0047]
室温下将5.00kg中间体ⅰ加入78.75kg冰醋酸中,氮气置换后,加入9.47kg三溴化吡啶鎓。加毕,升至35~45℃搅拌2h。反应完成后,将反应液降温至15~25℃,缓慢加入75.00kg水。加毕,离心,滤饼用水淋洗。减压干燥得到6.88kg 5-(2-溴乙酰)噻吩-2-甲酰胺,收率93.84%,hplc纯度94.02%。
[0048]1h-nmr(dmso-d6,δtms0):4.67ppm(2h,s),7.69ppm(1h,s),7.85ppm(1h,d,j=
3.6),8.21ppm(1h,s),8.18ppm(1h,d,j=4.0)
[0049]
实施例2:5-(2-溴乙酰)噻吩-2-甲酰胺的制备
[0050]
(a)中间体ⅰ的制备:
[0051]
将115g5-乙酰-噻吩-2-羧酸悬浮于1524g乙腈中,加入2.3gn,n-二甲基甲酰胺,氮气置换后搅拌降温至0℃,缓慢加入128.9g草酰氯。加毕,缓慢升温至30~40℃搅拌。反应完全后,将反应液减压浓缩至干。残余物加入1524g乙腈,搅拌溶解完全后,备用。向反应釜中加入1.15l浓氨水,降温至15~25℃,缓慢加入上述中间态——酰氯的乙腈溶液。加毕,过滤,滤饼用水淋洗。减压干燥得到108.8kg中间体ⅰ,收率95.15%,hplc纯度98.78%。
[0052]
(b)5-(2-溴乙酰)噻吩-2-甲酰胺的制备:
[0053]
室温下将20.0g中间体ⅰ加入210g冰醋酸中,氮气置换后,加入48.4g四溴环酮。加毕,升至30~40℃搅拌4h。反应完成后,将反应液降温至15~25℃,缓慢加入200.0g水。加毕,过滤,滤饼用水淋洗。减压干燥得到28.2g 5-(2-溴乙酰)噻吩-2-甲酰胺,收率96.15%,hplc纯度91.29%。
[0054]
实施例3:5-(2-溴乙酰)噻吩-2-甲酰胺的制备
[0055]
(a)中间体ⅰ的制备:
[0056]
将115g5-乙酰-噻吩-2-羧酸悬浮于1524g四氢呋喃中,加入2.3gn,n-二甲基甲酰胺,氮气置换后搅拌降温至0℃,缓慢加入94.4g草酰氯。加毕,缓慢升温至10~15℃搅拌。反应完全后,将反应液减压浓缩至干。残余物加入1524g四氢呋喃,搅拌溶解完全后,备用。向反应釜中加入172.5ml浓氨水,降温至-10~0℃,缓慢加入上述中间态——酰氯的四氢呋喃溶液。加毕,过滤,滤饼用水淋洗。减压干燥得到110.1kg中间体ⅰ,收率96.29%,hplc纯度98.40%。
[0057]
(b)5-(2-溴乙酰)噻吩-2-甲酰胺的制备:
[0058]
室温下将20.0kg中间体ⅰ加入252g冰醋酸中,氮气置换后,加入51.3g四丁基三溴化铵。加毕,升至40~50℃搅拌2h。反应完成后,将反应液降温至15~25℃,缓慢加入240.0g水。加毕,过滤,滤饼用水淋洗。减压干燥得到26.7g 5-(2-溴乙酰)噻吩-2-甲酰胺,收率91.03%,hplc纯度92.43%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1