一种维生素A杂油的提纯方法与流程
一种维生素a杂油的提纯方法
技术领域
1.本发明涉有机化合物提纯领域,具体地说是涉及一种维生素a杂油的提纯方法。
背景技术:
2.维生素a(简称va)是一种脂溶性维生素,是人体和动物必须维生素之一。在医药、食品、饲料添加剂及化妆品等行业都具有重要的应用。具体结构如下:
[0003][0004]
维生素a虽然可以从动物组织中提取,但资源相对分散,步骤繁杂,成本较高,因此商品维生素a都是化学合成产品。目前主要的代表合成路线有:1)罗氏公司的c6+c14路线;2)巴斯夫公司的c5+c15路线。维生素a粗品为油状液体,一般将其溶解于乙醇中通过低温结晶得到纯度大于等于270万iu的维生素a晶体。结晶母液脱除溶剂后得到的油状液体称为va杂油。va杂油中va含量一般为50~100万iu,如何进一步利用杂油中的va是降低生产成本、提高市场竞争力的关键。根据目前相关产品的市场价格及毛利率,最具经济效益的方式是对va杂油再次进行结晶分离以得到更多的维生素a晶体产品,但由于va杂油中杂质含量较高,在正常结晶温度下va晶体很难析出,而当进一步降低结晶温度时,部分杂质会胶状化,阻碍结晶过程的正常进行。因此,对va杂油进行提纯处理十分重要。
[0005]
专利cn103936642中报道将va杂油依次通过正相萃取塔和反相萃取塔,通过高低两种极性溶剂对va的反复萃取,除掉母液中不同极性杂质进而提高va含量。此种提纯方法过程相对复杂,且需要使用专业的连续萃取设备,工艺操作复杂,成本投资较高。
[0006]
专利cn108218751中报道采用柱层析的方法将va与杂质进行分离。柱层析法常用于实验室对少量样品进行提纯分离定性使用,部分制药企业也将其用于高附加值药品的分离,但产量较低,年产量一般不超过100kg。对于大规模工业化产品,柱层析显然不适用。
[0007]
专利cn113292467中报道采用无机碱的醇水溶液对va杂油萃取洗涤,在一定ph值范围内将母液中的杂质萃取到醇水溶液中,从而实现va杂油的提纯。该方法虽然操作简便,但其提纯效果及va回收率均相对较低,根据专利中实施例报道,提纯后的va精油中va最高含量仅为207万iu,va最高回收率仅为81%左右。且该工艺会产生大量的含有碱性无机盐废水,增加生产成本。
技术实现要素:
[0008]
为了解决以上技术问题,本发明提供了一种va杂油的提纯方法。通过该方法可以将杂油中va含量提高至230万iu以上且va回收率高达90%以上。
[0009]
为实现上述目的,本发明所采用的技术方案如下:
[0010]
一种维生素a杂油的提纯方法,所述方法包含以下步骤;
[0011]
s1:将有机碱溶于乙腈,并入助剂,得到有机碱的乙腈溶液;
[0012]
s2:在维生素a杂油中加入烷烃溶剂混合均匀后,加入有机碱的乙腈溶液,充分混合,得到混合溶液;
[0013]
s3:向混合溶液中加入水,充分搅拌后静置分相,分去下层乙腈水相,脱除溶剂,得到提纯的维生素a精油。
[0014]
所述的提纯过程是在有机碱体系中进行,有助于维生素a杂油中的杂质溶解在乙腈水体系中,所添加的芳香族醚类助剂可以提高杂质在乙腈水体系中的分配系数,从而提高提纯效果。
[0015]
本发明中,s1所述的有机碱为含有c1~c4饱和脂肪族的醇胺类化合物,优选为乙醇胺、二乙醇胺、三乙醇胺、异丙醇胺、三异丙醇胺中的一种或多种;优选地,所述有机碱的乙腈溶液浓度优选为1~10wt%,优选3~8wt%。
[0016]
本发明中,s1所述助剂为芳香族醚化合物,优选2-氟苯甲醚、3-氟苯甲醚、4-氟苯甲醚、3-氯苯甲醚、4-氯苯甲醚中的一种或多种;优选地,所述助剂与有机碱的质量比为(0.2~1.0):1,优选为(0.4~0.8):1。
[0017]
本发明中,s2所述烷烃溶剂为c5~c18的直链烷烃、c5~c18的支链烷烃和c5~c18的环烷烃,优选为正戊烷、正己烷、正庚烷中的一种或多种;优选地,所述烷烃溶剂与维生素a杂油的质量比为(1~9):1,优选为(1.5~4):1。
[0018]
本发明中,s2所述有机碱的乙腈溶液与维生素a杂油的质量比为(1~5):1,优选为(2~4):1。
[0019]
本发明中,所述s2加入有机碱的乙腈溶液后在-5~25℃下搅拌30~120min,优选在5~15℃下搅拌60~90min。
[0020]
本发明中,所述s3加入的水与有机碱的乙腈溶液的质量比为(0.1~0.5):1,优选为(0.2~0.4):1。
[0021]
本发明中,所述s3脱除溶剂采用减压蒸馏,条件为温度30~50℃,真空度5~160kpaa。
[0022]
本发明的另一目的在于提供一种提纯方法的用途。
[0023]
一种提纯方法的用途,上述的提纯方法用于维生素a杂油的提纯。
[0024]
与现有技术相比,本发明具有如下有益效果:
[0025]
(1)本发明工艺提纯后va精油中va含量高达230万iu以上,va回收率高达90%以上;
[0026]
(2)本发明工艺提纯后的va精油可再次进行结晶分离获得va晶体,最大化提高产品收率,提高产品市场竞争力;
[0027]
(3)本发明工艺操作流程简便,设备成本投资低,不需要价格高昂的专业萃取设备,适用于规模化生产。
具体实施方式
[0028]
下面通过具体实施例对本发明做进一步说明,本发明所述实施例只是作为对本发明的说明,不限制本发明的范围。
[0029]
(1)液相色谱分析条件:
[0030]
高效液相色谱仪,安捷伦lc-1200,色谱分析条件:按照gb 14750-2010规定的条件进行测定。维生素a含量通过外标法进行测定。
[0031]
(2)本发明各实施例中有机碱来源如下:
[0032]
乙醇胺:ar 99%,阿拉丁
[0033]
二乙醇胺:ar 99%,阿拉丁
[0034]
三乙醇胺:≥99%(gc),阿拉丁
[0035]
异丙醇胺:>93%(gc),阿拉丁
[0036]
三异丙醇胺:95%,异构体混合物,阿拉丁
[0037]
丁醇胺:97%,阿拉丁
[0038]
n-丁基二乙醇胺:98%,阿拉丁
[0039]
2-氟苯甲醚:98%,阿拉丁
[0040]
3-氟苯甲醚:99%,阿拉丁
[0041]
4-氟苯甲醚:99%,阿拉丁
[0042]
3-氯苯甲醚:98%,阿拉丁
[0043]
4-氯苯甲醚:99%,阿拉丁
[0044]
(3)本发明各实施例及对比例采用的va杂油来源及组成:
[0045]
杂油a:来自于万华化学维生素a合成的小试工艺,杂油中va含量为53万iu,其余为重组分杂质。
[0046]
杂油b:来自于万华化学维生素a合成的小试工艺,杂油中va含量为79万iu,其余为重组分杂质。
[0047]
杂油c:来自于万华化学维生素a合成的小试工艺,杂油中va含量为97万iu,其余为重组分杂质。
[0048]
其他原料及试剂若无特殊说明,均通过市售商业途径购买获得。
[0049]
(4)本发明各实施例中所使用减压蒸馏设备信息如下:
[0050]
真空系统:采用knf系列sc 920g真空泵;
[0051]
蒸馏设备:欣维尔d54102型微量垂刺蒸馏器。
[0052]
实施例1
[0053]
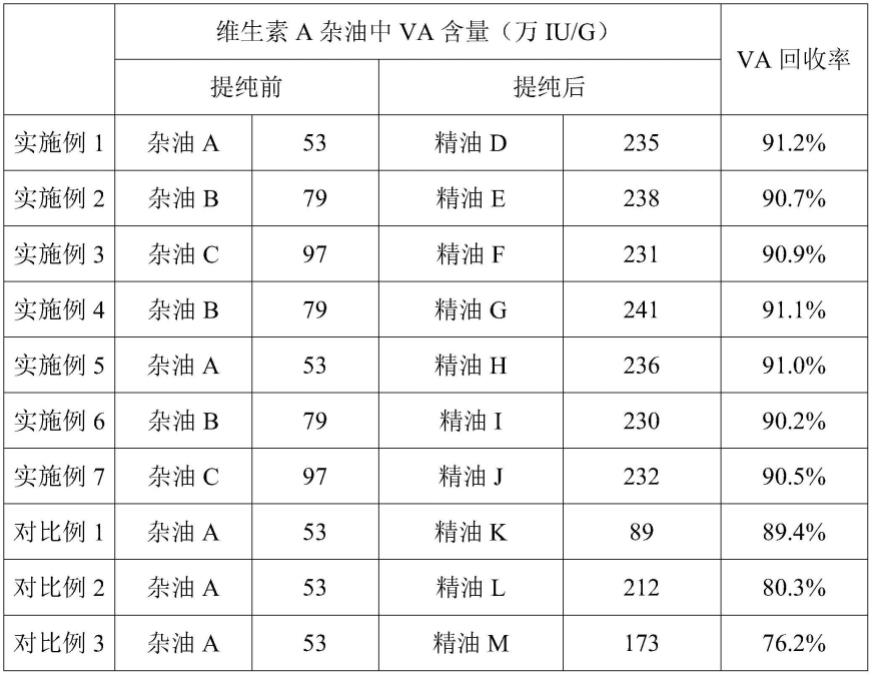
称取7.5g三乙醇胺溶解于142.5g乙腈中配制成浓度5%的三乙醇胺的乙腈溶液,并称取4.5g的2-氟苯甲醚加入到上述溶液中搅拌至完全溶解;称取50.0g杂油a溶解于125.0g正己烷中;将乙腈溶液加入到烷烃溶液中并在10℃下充分搅拌80min;向混合溶液中加入45.0g纯水,继续充分搅拌5min后静置分相;分去下层乙腈水相,在40℃,真空度37kpaa下减压蒸馏脱出溶剂得到提纯后的精油d10.3g,通过高效液相色谱对精油d中va含量进行分析,结果如表1所示。
[0054]
实施例2
[0055]
称取1.8g乙醇胺溶解于58.2g乙腈中配制成浓度3%的乙醇胺的乙腈溶液,并称取1.8g的3-氟苯甲醚加入到上述溶液中搅拌至完全溶解;称取30.0gva杂油b溶解于120.0g正戊烷中;将乙腈溶液加入到烷烃溶液中并在15℃下充分搅拌60min;向混合溶液中加入12.0g纯水,继续充分搅拌5min后静置分相;分去下层乙腈水相,在30℃,真空度82kpaa下减压蒸馏脱出溶剂正戊烷得到提纯后的精油e 9.0g,通过高效液相色谱对精油e中va含量进
行分析,结果如表1所示。
[0056]
实施例3
[0057]
称取19.2g二乙醇胺溶解于220.8g乙腈中配制成浓度8%的二乙醇胺的乙腈溶液,并称取3.84g的4-氟苯甲醚加入到上述溶液中搅拌至完全溶解;称取60.0g杂油c溶解于90.0g正庚烷中;将乙腈溶液加入到烷烃溶液中并在5℃下充分搅拌90min;向混合溶液中加入96.0g纯水,继续充分搅拌5min后静置分相;分去下层乙腈水相,在40℃,真空度12kpaa下减压蒸馏脱出溶剂得到提纯后的精油f 22.9g,通过高效液相色谱对精油f中va含量进行分析,结果如表1所示。
[0058]
实施例4
[0059]
称取1.8g异丙醇胺溶解于178.2g乙腈中配制成浓度1%的异丙醇胺的乙腈溶液,并称取1.44g的3-氯苯甲醚加入到上述溶液中搅拌至完全溶解;称取36.0g杂油b溶解于324.0g正己烷中;将乙腈溶液加入到烷烃溶液中并在25℃下充分搅拌30min;向混合溶液中加入90.0g纯水,继续充分搅拌5min后静置分相;分去下层乙腈水相,在50℃,真空度54kpaa下减压蒸馏脱出溶剂得到提纯后的精油g10.8g,通过高效液相色谱对精油g中va含量进行分析,结果如表1所示。
[0060]
实施例5
[0061]
称取9.0g三异丙醇胺溶解于81.0g乙腈中配制成浓度10%的三异丙醇胺的乙腈溶液,并称取3.6g的4-氯苯甲醚加入到上述溶液中搅拌至完全溶解;称取90.0gva杂油a溶解于90.0g正戊烷中;将乙腈溶液加入到烷烃溶液中并在-5℃下充分搅拌120min;向混合溶液中加入9.0g纯水,继续充分搅拌5min后静置分相;分去下层乙腈水相,在40℃,真空度115kpaa下减压蒸馏脱出溶剂得到提纯后的精油h18.4g,通过高效液相色谱对精油h中va含量进行分析,结果如表1所示。
[0062]
实施例6
[0063]
称取5.4g丁醇胺溶解于54.60g乙腈中配制成浓度9%的丁醇胺的乙腈溶液,并称取1.62g的2,4,6-三氟苯甲醚加入到上述溶液中搅拌至完全溶解;称取40.0g杂油b溶解于200.0g正己烷中;将乙腈溶液加入到烷烃溶液中并在0℃下充分搅拌110min;向混合溶液中加入9.0g纯水,继续充分搅拌5min后静置分相;分去下层乙腈水相,在40℃,真空度37kpaa下减压蒸馏脱出溶剂得到提纯后的精油i12.4g,通过高效液相色谱对精油i中va含量进行分析,结果如表1所示。
[0064]
实施例7
[0065]
称取6.3g n-丁基二乙醇胺溶解于308.7g乙腈中配制成浓度2%的n-丁基二乙醇胺的乙腈溶液,并称取5.67g的3-溴-4-氯苯甲醚加入到上述溶液中搅拌至完全溶解;称取70.0g杂油c溶解于490.0g正己烷中;将乙腈溶液加入到烷烃溶液中并在20℃下充分搅拌50min;向混合溶液中加入141.75g纯水,继续充分搅拌5min后静置分相;分去下层乙腈水相,在40℃,真空度37kpaa下减压蒸馏脱出溶剂得到提纯后的精油j 26.5g,通过高效液相色谱对精油j中va含量进行分析,结果如表1所示。
[0066]
对比例1
[0067]
称取4.5g的2-氟苯甲醚加入到150g乙腈中搅拌至完全溶解;称取50.0g杂油a溶解于125.0g正己烷中;将乙腈溶液加入到烷烃溶液中并在10℃下充分搅拌80min;向混合溶液
中加入45.0g纯水,继续充分搅拌5min后静置分相;分去下层乙腈水相,在40℃,真空度37kpaa下减压蒸馏脱出溶剂得到提纯后的精油k 26.6g,通过高效液相色谱对精油k中va含量进行分析,结果如表1所示。
[0068]
对比例2
[0069]
称取7.5g三乙醇胺溶解于142.5g乙腈中配制成浓度5%的三乙醇胺的乙腈溶液;称取50.0g杂油a溶解于125.0g正己烷中;将乙腈溶液加入到烷烃溶液中并在10℃下充分搅拌80min;向混合溶液中加入45.0g纯水,继续充分搅拌5min后静置分相;分去下层乙腈水相,在40℃,真空度37kpaa下减压蒸馏脱出溶剂得到提纯后的精油l11.3g,通过高效液相色谱对精油l中va含量进行分析,结果如表1所示。
[0070]
对比例3
[0071]
称取7.5g氢氧化钠溶解于67.5g水中配制成浓度10%的氢氧化钠水溶液;称取5.0g水溶解于45.0g乙醇中配制成浓度10%的水的乙醇溶液;称取50.0g杂油a溶解于100.0g正己烷中;将上述氢氧化钠水溶液和乙醇水溶液加入到正己烷溶液中,在30℃下充分搅拌80min后静置分相,分去下层水相,加50g水洗涤一遍,继续分水操作;将正己烷相在40℃,真空度37kpaa下减压蒸馏脱出溶剂得到提纯后的精油m11.7g,通过高效液相色谱对精油m中va含量进行分析,结果如表1所示。
[0072]
表1实施例1~7和对比例1~3结果
[0073]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1