一种联噻吩与噻吩基异靛青交替共轭聚合物、其制备方法和应用
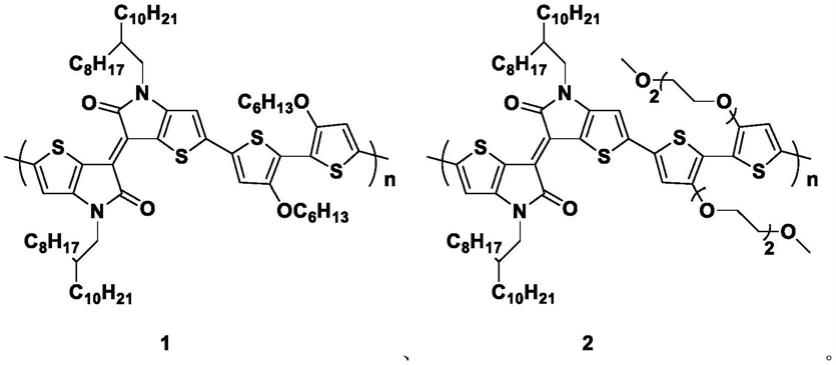
1.本发明涉及有机热电材料技术领域,尤其涉及一种联噻吩与噻吩基异靛青交替共轭聚合物、其制备方法和应用。
背景技术:
2.热电材料在解决能量转换过程中产生废热问题上是一种非常理想的途径,它是一种可以将热能直接转换为电能的材料。相比于其他能量转换材料,热电材料具有无机械传动装置、无工作噪声和气体污染、器件结构简单轻便易微型化、使用环境不受限制及工作寿命长等优点,在航空航天、医疗器械及可穿戴设备等领域都有非常好的应用前景
3.给受体(d-a)共轭聚合物一般指给体和受体单元交替共聚所形成的均匀结构,其最高占有分子轨道(homo)和最低未占分子轨道(lumo)分别由给体单元、受体单元决定。d-a共轭聚合物分子内发生电荷转移,分子轨道混合导致带隙eg较窄,载流子迁移率高。
4.噻吩基异靛青(tiig)中氧和硫元素相互作用使得分子结构平面性优异,基于tiig的聚合物具有较高的载流子迁移率,有利于在较低掺杂浓度下实现较高的电导率。然而这些共轭聚合物通常在常见溶剂中的溶解性较差,与掺杂剂的共混性不好。
技术实现要素:
5.本发明为了解决上述技术问题提供一种联噻吩与噻吩基异靛青交替共轭聚合物。本发明引入烷氧、醚氧侧链提高其homo能级,进而通过混合掺杂可使其具有较高的电导率和塞贝克系数,具有良好的溶解性以及与掺杂剂的共混性,经过掺杂后具有较高的电导率,具有更好的热电性能。
6.本发明解决上述技术问题的技术方案如下:
7.一种联噻吩与噻吩基异靛青交替共轭聚合物,其化学结构式如式i所示:
[0008][0009]
其中,r1表示为烷基;
[0010]
r2表示为-oc6h
13
和-och2ch2och2ch2och3中的任意一种;
[0011]
n表示为19~24的整数。
[0012]
采用上述方案的有益效果是:
[0013]
研究发现,本发明的联噻吩与噻吩基异靛青交替共轭聚合物引入烷氧、醚氧侧链提高其homo能级,具有良好的溶解性以及与掺杂剂的共混性,经过掺杂后具有较高的电导
率,具有更好的热电性能。
[0014]
在上述技术方案的基础上,本发明还可以做如下改进。
[0015]
进一步的,所述r1表示为直链烷基和支链烷基中的任意一种,所述直链烷基表示为含有6~12个碳原子的直链烷基链,所述支链烷基表示为支链含有6~12个碳原子和主链含有6~12个碳原子的支链烷基链。
[0016]
采用上述进一步方案的有益效果是:r1采用支链烷基和支链烷基能够提升掺杂后的导电率和热电性能。
[0017]
进一步的,所述联噻吩与噻吩基异靛青交替共轭聚合物为如下具体结构式中的任意一种:
[0018][0019]
采用上述进一步方案的有益效果是:上述结构式的聚合物与掺杂剂掺杂后电导率和热电性能最佳。
[0020]
本发明还提供上述的联噻吩与噻吩基异靛青交替共轭聚合物的制备方法,反应式如下:
[0021]
[0022][0023]
其中,r1表示为烷基;
[0024]
r2表示为-oc6h
13
和-och2ch2och2ch2och3中的任意一种;
[0025]
n表示为19~24的整数;
[0026]
制备包括以下步骤:
[0027]
步骤a:如反应式一所示,将原料a、n-buli和fe(acac)3在thf中催化反应,制得中间体a;
[0028]
步骤b:如反应式二所示,将中间体a、n-buli和(ch3)3sncl在thf中反应,制得中间体b;
[0029]
步骤c:如反应式三所示,将原料b、中间体b和pd(pph3)4在甲苯中反应,制得式i所示的化合物,即为联噻吩与噻吩基异靛青交替共轭聚合物。
[0030]
采用上述制备方法的有益效果是:
[0031]
上述制备方法简单可行,能够制得电导率和热电能更好的聚合物。
[0032]
在上述技术方案的基础上,本发明还可以做如下改进。
[0033]
进一步的,步骤a中,所述原料a、所述n-buli和所述fe(acac)3反应的摩尔比为1:(1~2):(1~1.8),反应温度为-20~75℃。
[0034]
采用上述进一步方案的有益效果是:步骤a采用上述材料制得中间体a的产率高。
[0035]
进一步的,步骤b中,所述中间体、所述n-buli和所述(ch3)3sncl反应的摩尔比为1:(1~1.5):(1~1.5),反应温度为-78~80℃。
[0036]
采用上述进一步方案的有益效果是:步骤b采用上述材料制得的中间体b的产率高。
[0037]
进一步的,步骤c中,所述原料b、所述中间体b和所述pd(pph3)4反应的摩尔比为1:(0.9~1.1):(0.01~0.04),反应温度为105~115℃。
[0038]
采用上述进一步方案的有益效果是:采用上述材料和反应温度,聚合物的产率高。
[0039]
本发明还提供上述的联噻吩与噻吩基异靛青交替共轭聚合物在制备热电材料中的应用。
[0040]
上述应用的有益效果是:
[0041]
通过引入强受体单元噻吩基异靛青以及含有不同侧链联噻吩衍生物给体单元的聚合物,可与掺杂剂合成高效热电材料;通过改变给体单元侧链结构,同时对聚合物的掺杂条件进行优化,提高有机热电材料的热电性能。
[0042]
在上述技术方案的基础上,本发明还可以做如下改进。
[0043]
进一步的,所述热电材料由权利要求1-3任一项所述的联噻吩与噻吩基异靛青交替共轭聚合物和掺杂剂按摩尔比为10:(1~5)制成。
[0044]
进一步的,所述掺杂剂为2,3,5,6-四氟-7,7',8,8'-四氰二甲基对苯醌。
[0045]
采用上述进一步方案的有益效果是:采用上述掺杂剂,能够和联噻吩与噻吩基异靛青交替共轭聚合物共混,所得的热电材料具有更好的电导率和热电性能。
附图说明
[0046]
图1为本发明实施例3的聚合物ptiig-btor掺杂前后聚合物薄膜的紫外可见近红外吸收光谱图;
[0047]
图2为本发明实施例3的聚合物ptiig-bt2egome掺杂前后聚合物薄膜的紫外可见近红外吸收光谱图。
具体实施方式
[0048]
以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
[0049]
实施例1
[0050]
聚合物ptiig-btor:
[0051]
[0052]
聚合物ptiig-btor的制备路线如下:
[0053][0054][0055]
聚合物ptiig-btor的制备,具体为:
[0056]
1、将100ml双口瓶烘干后加入磁子,抽换气使反应气氛为n2,注入2.7116g原料1a和15ml无水无氧四氢呋喃,置于冰水浴15min,逐滴加6.0ml正丁基锂反应,反应液由无色透明变为浅黄色,然后在冰盐浴反应2h,得浅黄色反应液;将另一个100ml双口瓶烘干后加入磁子,n2抽换气冷却,加入5.1988g三乙酰丙酮铁,再抽换气使反应气氛为n2,注入30ml无水无氧四氢呋喃,然后将之前所得浅黄色反应液用经导针转移至反应瓶中,40℃油浴下反应3h,于68℃油浴回流反应过夜;tlc检测反应完毕后,用二氯甲烷溶解反应液,漏斗内加入少量硅胶粉对反应液抽滤,得到滤液,滤液为红黑色液体;对滤液进行减压旋蒸浓缩后,硅胶柱层析(洗脱剂为石油醚:二氯甲烷=10:1)纯化得到黄色固体,将黄色固体用石油醚重结晶三次得到中间体1a,中间体1a淡绿色雪花状晶体;
[0057]
2、将100ml双口瓶烘干后加入磁子,抽换气使反应气氛为n2,加入1.1g中间体1a,再次抽换气使反应气氛为n2,用注射器注入40ml无水无氧四氢呋喃,低温浴冷却至-78℃,然后逐滴滴加2.7ml正丁基锂,反应液由透明变为乳白色液体,反应时有固体析出时,补加无水无氧四氢呋喃,正丁基锂滴加完成后搅拌反应2h,反应液变为乳黄色,升至室温,n2保护下直接加入1.3198g三甲基氯化锡,室温反应过夜;tlc检测反应完毕后,用去离子水淬灭反应瓶中剩余正丁基锂,然后用二氯甲烷萃取三次,收集有机相水洗两次,用无水na2so4干燥,抽滤,滤液减压旋蒸,中性氧化铝柱层析(洗脱剂为石油醚:三乙胺=97:3)纯化得到橙红色片状初产物,再用无水乙醇重结晶三次,烘干,得到1.0852g中间体1b,中间体1b为橙红色片状晶体,产率52.2%;
[0058]
3、将50ml聚合管烘干后抽换气并冷却排尽水汽,加入248.3mg原料b、173.0mg中间体1b、5.9mg的四(三苯基膦)钯以及磁子,再次抽换气处理使反应气氛为n2,注入8ml无水无氧的甲苯,在110℃油浴反应96h,反应完毕冷却至室温后,将反应液滴加于搅拌中的200ml
无水甲醇中,沉降,抽滤,滤饼烘干,用定性滤纸包好放入索氏提取器中,依次使用丙酮、正己烷以及三氯甲烷进行抽提,分别收集溶解于正己烷、三氯甲烷中的聚合物溶液,聚合物溶液减压旋蒸浓缩,再次在甲醇中沉降,抽滤,滤饼烘干后得到共258.2mg化合物1;其中,正己烷抽提得到49mg聚合物ptiig-btor,三氯甲烷抽提得到209.2mg聚合物ptiig-btor,合并产率86.1%。
[0059]
实施例2
[0060]
聚合物ptiig-bt2egome:
[0061][0062]
聚合物ptiig-bt2egome的制备路线如下:
[0063][0064]
聚合物ptiig-bt2egome的制备,具体为:
[0065]
1、将100ml双口瓶烘干后加入磁子,抽换气使反应气氛为n2,注入6.0678g原料2a和30ml无水无氧四氢呋喃,置于冰水浴15min,逐滴加12.0ml正丁基锂反应,反应液由无色
透明变为浅黄色,然后在冰盐浴反应2h,得到浅黄色反应液;将另一个100ml双口瓶烘干后加入磁子,n2抽换气冷却,加入10.601g三乙酰丙酮铁,再抽换气使反应气氛为n2,注入40ml无水无氧四氢呋喃,然后将之前所得浅黄色反应液用经导针转移至反应瓶中,40℃油浴反应3h,于68℃油浴回流反应过夜;tlc检测反应完毕后,用二氯甲烷溶解反应液,漏斗内加入少量硅胶粉对反应液抽滤,得到滤液,滤液减压旋蒸浓缩,硅胶柱层析(洗脱剂为石油醚:乙酸乙酯=4:1)纯化得到中间体2a;
[0066]
2、将50ml双口瓶烘干后抽换气并冷却排尽水汽,先加入1.2146g中间体2a及磁子,再次抽换气使反应气氛为n2,注入40ml无水无氧四氢呋喃,低温浴冷却至-78℃,逐滴加入2.7ml正丁基锂,反应液由黄绿色变为橙黄色,反应有固体析出时,补加无水无氧四氢呋喃,正丁基锂滴加完成后搅拌反应2h,反应液变为乳黄色,升至室温,n2保护下直接加入1.3154g三甲基氯化锡,室温反应过夜;tlc检测反应完毕后,用去离子水淬灭反应瓶中剩余正丁基锂,乙酸乙酯萃取三次,收集有机相水洗两次,用无水na2so4干燥,抽滤,滤液减压旋蒸,中性氧化铝柱层析(洗脱剂为石油醚:乙酸乙酯=4:1)分离得到黄白色针状初产物,再用无水乙醇重结晶三次,烘干,得到1.0344g中间体2b,产率47.4%;
[0067]
3、将25ml聚合管烘干后抽换气并冷却排尽水汽,加入248.4mg原料b、182.2mg中间体2b、6.1mg的四(三苯基膦)钯以及磁子,再次抽换气处理使反应气氛为n2,注入8ml无水无氧的甲苯,110℃油浴反应96h,反应完毕冷却至室温后,将反应液滴加于搅拌中的200ml无水甲醇中,沉降,抽滤,滤饼烘干,用定性滤纸包好放入索氏提取器中,依次使用丙酮、正己烷以及三氯甲烷进行抽提,收集溶解于三氯甲烷中的聚合物溶液,减压旋蒸浓缩,再次在甲醇中沉降,抽滤,滤饼烘干,得到267.7mg聚合物ptiig-bt2egome,产率86.6%,聚合物ptiig-bt2egome为黑色固体。
[0068]
实验例
[0069]
以实施例1和实施例2制备的聚合物为有机热电材料进行热电性能测试。
[0070]
以1.25cm
×
1.25cm载玻片为基底,使用前用超纯水、丙酮和异丙醇清洗然后用紫外臭氧清洗仪处理15min,随后将二氯甲烷溶解的聚合物溶液和乙腈溶解的f4tcnq溶液混合,f4tcnq的掺杂量为10%到50%摩尔比,得到混合液;取80μl混合液均匀滴覆在基地上,采用旋涂仪(转速1500rpm到4000rpm)旋涂成膜,旋膜完成后在50℃热台下干燥10分钟,即得聚合物薄膜。如图1、2所示,通过薄膜的紫外可见吸收光谱可以看到,经过掺杂后,聚合物薄膜在900-1100的中性吸收峰强度降低,在1400以上形成了新吸收峰,说明采用共混掺杂的方式可以对聚合物1和聚合物2进行有效的氧化掺杂。
[0071]
通过四探针法测试掺杂后的聚合物薄膜的方块电阻,用台阶仪测试薄膜厚度,从而可获得聚合物薄膜的电导率。利用薄膜热电参数测试系统可获得聚合物薄膜的塞贝克系数。
[0072]
通过掺杂剂浓度的优化和掺杂条件的选择,经过大量的实验测试可得ptiig-btor、ptiig-bt2egome热电数据,分别如表1、表2所示:
[0073]
表1:
[0074][0075]
表2:
[0076][0077]
由表1和表2可以看出,聚合物随着掺杂浓度的提升电导率逐渐增大,塞贝克系数逐渐降低,ptiig-btor在30%掺杂浓度下达到最优热电性能2.76μw m-1
k-2
,ptiig-bt2egome在40%掺杂浓度下达到最优热电性能4.99μw m-1
k-2
,其热电参数变化规律与文献
[1]
报道一致。以上数据表明本发明的联噻吩与噻吩基异靛青交替共轭聚合物在热电材料中有很大的发展潜力。
[0078]
参考文献:
[0079]
[1]j.mater.chem.c,2021,9,340;int.j.energ.res.,2021,45,21540.
[0080]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1