一种骨化三醇有关杂质PZB的分离纯化方法与流程
一种骨化三醇有关杂质pzb的分离纯化方法
技术领域
1.本发明属于化学制药技术领域,具体涉及一种骨化三醇的有关杂质pzb的分离纯化方法以及利用该杂质作为杂质对照品,在骨化三醇原料药和制剂中的检测与分析等质量控制方面的用途。
背景技术:
2.骨化三醇(calcitriol),化学名为:1,25-二羟胆钙化醇或1,25-二羟维生素d3,结构式为:
[0003][0004]
骨化三醇是维生素d3经肝脏和肾脏羟化酶代谢为抗佝偻病活性最强的代谢物。能促进肠道钙的吸收并调节骨质的钙化。骨化三醇最早由瑞士罗氏公司生产,1978年上市,商品名为“rocaltrol”,用于治疗绝经后骨质疏松、慢性肾功能低下、术后甲状腺功能低下、特发性甲状旁腺功能低下、假性甲状腺功能低下、维生素d依赖性佝偻病、低血磷性维生素d抵抗型佝偻病等。由于骨化三醇是内源性物质,疗效确切、安全稳定,因此是骨质疏松症的首选药物。
[0005]
骨化三醇生理活性很高,单次服用剂量极小,一般治疗剂量仅0.25μg/日-1.0μg/日。骨化三醇性质不稳定,对光和热敏感,含有多个手性中心,传统合成路线较长,在合成过程中无可避免的容易产生一些异构体杂质及其它有关物质,难于分离纯化,因而合成工艺技术门槛高、难度大。因此,对原料药和制剂产品的质量控制提出了更高的要求。
[0006]
目前,中国药典(2020版)未收载骨化三醇品种,欧洲药典(ep9.0)及美国药典(usp40)中收载了该品种,并控制了杂质包括:骨化三醇前体、反式骨化三醇、1β-骨化三醇、亚甲基骨化三醇以及骨化三醇前体三唑啉加合物。除此之外,专利cn112552265a公开了一种骨化三醇杂质,属于光反应合成过程中产生的氧化降解杂质。专利cn111978230a公开了一种骨化三醇类化合物,是通过强降解实验产生的杂质。
[0007]
本技术骨化三醇的制备工艺区别于传统光化学合成法,是采用微生物发酵法制得,在发酵液提取纯化过程中,意外发现一个含量极高的杂质pzb,含量高达7%以上,且薄层点板发现杂质极位点与骨化三醇相近,采用柱层析、dac动态制备等纯化方法难以去除。经分离、鉴定和结构确认,该杂质并未被收入上述药典或标准中。因此,确定该杂质的化学结构和制备方法,建立检测方法,分析杂质含量,并确定合理的杂质限度,对保证骨化三醇的产品质量及用药安全性十分必要。
技术实现要素:
[0008]
为了解决上述问题,本发明提供一种骨化三醇的有关杂质pzb的分离纯化方法。
[0009]
实现本发明的技术方案是:
[0010]
本发明提供一种骨化三醇的有关杂质pzb的分离纯化方法,杂质pzb为式1所示的化合物:
[0011][0012]
所述分离纯化方法包括以下步骤:
[0013]
(1)发酵工艺:以阿法骨化醇为起始物料,经一步微生物发酵转化成骨化三醇,发酵液提取得到含有式1化合物的骨化三醇粗品;
[0014]
(2)tbs保护羟基:含有式1的骨化三醇粗品使用有机溶剂溶解,向其中加入tbscl、咪唑,室温下反应,tlc监测至原料反应完毕,再经柱层析分离,减压浓缩至干得到式3化合物;
[0015]
(3)脱tbs:式3化合物使用有机溶剂溶解,向其中加入tbaf
·
thf升温至50-60℃进行反应,tlc监测至原料反应完毕,经萃取、柱层析纯化、减压浓缩、烘干后得到白色固体,即式1化合物。
[0016]
具体反应式如下:
[0017]
[0018]
在一些实施方式中,步骤(1)中微生物发酵转化方法是将假诺卡氏菌hrw001进行发酵培养、离心分离后得到将假诺卡氏菌hrw001菌体,并将所述菌体应用于微生物转化式2化合物即起始物料阿法骨化醇。
[0019]
在一些实施方式中,步骤(1)中骨化三醇粗品中式1化合物含量为1~10%,优选为2~8%,进一步优选为5~8%。
[0020]
在一些实施方式中,步骤(2)中有机溶剂选自二氯甲烷、氯仿、苯、甲苯中的一种或几种。
[0021]
在一些实施方式中,步骤(3)中有机溶剂选自甲醇、乙醇、正丙醇、异丙醇丙酮、四氢呋喃、二甲醚、乙酸中的一种或几种。
[0022]
在一些实施方式中,步骤(2)中柱层析分离采用的洗脱剂为正己烷-乙酸乙酯溶液,具体方法为:正己烷润柱子,湿法上样,用正己烷:乙酸乙酯=80:1洗脱杂质,tlc监控,洗脱完毕后,将主点洗脱液于40~50℃减压浓缩干,得到式3化合物。
[0023]
在一些实施方式中,步骤(3)中柱层析纯化采用的洗脱剂为正己烷-乙酸乙酯溶液,具体方法为:正己烷润柱子,湿法上样,用正己烷:乙酸乙酯=5:1洗脱杂质,1:1分离主产物,tlc监控,洗脱完毕后,将主点洗脱液于40~50℃减压浓缩干,得到式1化合物。
[0024]
经本发明所述分离纯化方法制得的骨化三醇有关杂质pzb纯品的纯度大于98%,优选大于99%,最优选大于99.8%。
[0025]
与现有技术相比,本发明涉及的骨化三醇发酵杂质pzb及其制备方法、检测方法和用途的有益效果在于:
[0026]
(1)本发明提供了一种全新结构的骨化三醇的有关杂质pzb,经分离、鉴定和结构确认,该化合物是骨化三醇在微生物发酵过程中产生的微生物发酵杂质,该杂质在骨化三醇的原料发酵液中含量超过了7%,且经传统纯化方式难以去除,给产品安全性带来隐患。本发明提供了一种新的杂质制备方式,获得的杂质其高效液相纯度大于等于96.13%,对杂质对照品的制备具有重要的现实意义。
[0027]
(2)本发明通过分离获取该化合物,并对其进行结构确证从而确认该化合物的结构,并公开了hplc检测方法将其作为骨化三醇有关物质检查时的杂质对照品,从而有效的监控骨化三醇中的有关物质。本发明的实施有助于骨化三醇质量标准的提高,从而更好的控制骨化三醇的产品质量,对人民群众安全用药具有重要的意义。
附图说明
[0028]
图1为发酵上清液有关物质检测hplc图谱。
[0029]
图2为骨化三醇的有关杂质pzb的hplc图谱。
[0030]
图3为骨化三醇的有关杂质pzb的氢谱。
[0031]
图4为骨化三醇的有关杂质pzb的碳谱。
[0032]
图5为骨化三醇纯品有关物质检测hplc图谱。
具体实施方式
[0033]
下面结合具体实施例和附图对本发明作进一步详述,以下实施例只是描述性的,不是限定性的,不能以此限定本发明的保护范围,如无特别说明,所用原料均可通过市售或
自制获得。
[0034]
实施例1微生物转化制备骨化三醇粗品
[0035]
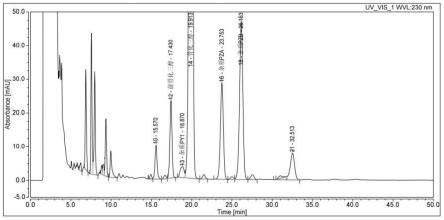
本发明所述骨化三醇采用微生物发酵法制得,具体制备方法参考专利公开号:cn110527650a中实施例1~5描述的方法获得,以阿法骨化醇为起始物料,经一步微生物发酵转化成骨化三醇粗品。微生物发酵采用的假诺卡氏菌hrw001。经中国普通微生物菌种保藏管理中心鉴定,分类命名为假诺卡氏菌pseudonocardia sp.hrw001,该菌株已于2019年04月03日保藏于中国微生物菌种保藏管理委员会普通微生物中心,保藏编号为cgmcc no.17524。发酵液经离心分离后检测发酵上清液有关物质hplc检测结果如表1,对应附图1。
[0036]
表1
[0037] 前骨化三醇杂质py1骨化三醇杂质pza杂质pzb阿法骨化醇出峰时间min17.43018.87019.91323.75326.15332.513峰面积比%2.480.6472.754.317.822.00
[0038]
以上结果表明,在发酵液提取纯化过程中,除了传统工艺杂质前骨化三醇和杂质py1(反式骨化三醇)外,还意外发现了两个含量极高的杂质pza和pzb,合计含量超过10%。
[0039]
本发明经经衍生化分离纯化得到这两个杂质,并经结构确证,其结构式及命名如下:
[0040]
杂质pza:
[0041]
中文化学名:(5z,7e)-9,10-开环胆甾-5,7,10(19)-三烯-1α,3β,26-三醇
[0042]
化学结构式:
[0043][0044]
杂质pzb:
[0045]
中文化学名:(5z,7e)-9,10-开环胆甾-5,7,10(19)-三烯-1α,3β,24-三醇
[0046]
化学结构式:
[0047][0048]
本发旨在保护杂质pzb的制备方法,建立检测方法,分析杂质含量,并确定合理的杂质限度,以保证骨化三醇的产品质量及用药安全性。
[0049]
从杂质pzb与骨化三醇的结构对比中,不难发现其主要结构差异在侧链引入羟基
位置的不同,其中杂质pzb的侧链24位羟基较易引入tbs保护基,而骨化三醇25位羟基的位阻较大,难以引入tbs保护基,因此,我们将发酵后所得的粗品,通过引入tbs保护基,利用骨化三醇只能1,3位羟基被tbs保护,而杂质pzb的三个羟基均能被tbs保护,故而通过tbs保护后两者极性大小的差异,利用柱层析将其分离,分离得到的杂质pzb的羟基tbs保护再经脱tbs保护,柱层析纯化,重结晶得到纯度较高的骨化三醇杂质pzb。
[0050]
实施例2杂质pzb的制备
[0051]
反应式如下:
[0052][0053]
制备方法如下:
[0054]
使用微生物转化的方法转化式2化合物即起始物料阿法骨化醇,提取得到含有式1化合物的骨化三醇粗品;
[0055]
将中试第一批经微生物转化提取所得的骨化三醇粗品(粗品中含有式1化合物)57.0g用570ml四氢呋喃溶解,加入2.0l反应瓶,向其中加入83.8g咪唑,92.9g的叔丁基二甲基氯硅烷,室温下反应2h,温度控制在20~30℃左右,tlc监测至原料反应完毕,减压浓缩至干,再经柱层析(正己烷润柱子,湿法上样,用正己烷:乙酸乙酯=80:1洗脱杂质)分离得到9.1g式3化合物;
[0056]
将9.1g式3化合物,用91ml四氢呋喃溶解,加入250ml反应瓶,加入54ml 1m tbaf
·
thf溶液,升温50-60℃反应3h,tlc监控,反应完全,减压浓缩至干,馏底物加入100ml乙酸乙酯和100ml水搅拌分层,有机相浓缩至干。浓缩物用50ml乙酸乙酯溶解,硅胶柱层析纯化(正己烷润柱子,湿法上样,用正己烷:乙酸乙酯=5:1洗脱杂质,1:1分离主产物),主产物洗脱剂浓缩至干,加入20ml乙酸乙酯、60ml石油醚搅洗0.5h,过滤,滤饼烘干得到3.16g。hplc纯度:96.56%。测定方法参见骨化三醇原料药质量标准有关物质检查方法;hplc谱图见2,核磁共振氢谱见图3、碳谱见图4,[m+h]
+
的m/z为417.33663,数据如表2表3所示。
[0057]
表2核磁共振氢谱测定结果
[0058]
[0059][0060]
表3核磁共振碳谱测定结果
[0061]
[0062]
[0063]
[0064][0065]
实施例3
[0066]
一种骨化三醇原料药质量标准有关物质检查方法,所述检测方法用于测定骨化三醇原料药中活性成分及有关物质的含量,具体包括以下步骤:
[0067]
(1)色谱条件设置:
[0068]
色谱柱:辛烷基硅烷键合硅胶为填充剂;
[0069]
柱温:38℃;
[0070]
流动相:1.0g/l三羟甲基氨基甲烷溶液-乙腈,乙腈体积百分浓度为45%;
[0071]
流速:2ml/min;
[0072]
检测波长:250nm;
[0073]
进样体积:50μl;
[0074]
运行时间:20min;
[0075]
(2)样品配制:
[0076]
供试品溶液制备:精密称取供试品适量,加入流动相溶解并定量稀释制成每1ml约含0.1mg的溶液;
[0077]
对照溶液制备:精密称取供试品溶液适量,用流动相定量稀释制成每1ml约含0.1μg的溶液;
[0078]
系统适用性溶液制备:另取骨化三醇对照品、杂质pzb对照品适量,加入流动相溶解并稀释制成每l ml中含骨化三醇0.1mg,式1化合物0.lμg的混合溶液,作为系统适用性溶液;
[0079]
(3)检测:
[0080]
精密量取所述供试品溶液和所述对照溶液50μl,分别注入液相色谱仪,记录色谱图至主峰保留时间的2倍,并采用自身对照法进行定量分析,计算骨化三醇及式1化合物的含量;系统适用性色谱图中,骨化三醇与式1化合物峰之间的分离度不小于1.5,理论板数以骨化三醇峰计算不低于10000。
[0081]
实施例4
[0082]
一种骨化三醇原料药质量标准有关物质检查方法,所述检测方法用于测定骨化三醇原料药中活性成分及有关物质的含量,具体包括以下步骤:
[0083]
(1)色谱条件设置:
[0084]
色谱柱:辛烷基硅烷键合硅胶为填充剂;
[0085]
柱温:40℃;
[0086]
流动相:1.0g/l三羟甲基氨基甲烷溶液-乙腈,乙腈体积百分浓度为55%;
[0087]
流速:1ml/min;
[0088]
检测波长:280nm;
[0089]
进样体积:50μl;
[0090]
运行时间:30min;
[0091]
(2)样品配制:
[0092]
供试品溶液制备:精密称取供试品适量,加入流动相溶解并定量稀释制成每1ml约含0.1mg的溶液;
[0093]
对照溶液制备:精密称取供试品溶液适量,用流动相定量稀释制成每1ml约含0.1μg的溶液;
[0094]
系统适用性溶液制备:另取骨化三醇对照品、杂质pzb对照品适量,加入流动相溶解并稀释制成每l ml中含骨化三醇0.1mg,式1化合物0.lμg的混合溶液,作为系统适用性溶液;
[0095]
(3)检测:
[0096]
精密量取所述供试品溶液和所述对照溶液50μl,分别注入液相色谱仪,记录色谱图至主峰保留时间的2倍,并采用自身对照法进行定量分析,计算骨化三醇及式1化合物的含量;系统适用性色谱图中,骨化三醇与式1化合物峰之间的分离度不小于1.5,理论板数以骨化三醇峰计算不低于10000。
[0097]
实施例5骨化三醇粗品制备
[0098]
将离心后的发酵液160l(以底物90g计)转移至500l反应釜内,加入25l乙酸乙酯,搅拌3~5min,静置3~5min,分液,有机层收集,水层舍去。有机层用1.8kg无水硫酸钠干燥,过滤,滤液40~50℃减压浓缩至干,向馏底物中加入0.45l正己烷于40℃下常压溶解待用。柱层析分离,正己烷润柱子,湿法上样,用正己烷:乙酸乙酯=80:1洗脱杂质,tlc监控(展开剂:正己烷/乙酸乙酯=1/2,显色方法:紫外254nm+磷钼酸加热),洗脱完毕后,将主点洗脱液于40~50℃减压浓缩干,得到57.0g油状物即为骨化三醇粗品。
[0099]
实施例6骨化三醇纯品制备
[0100]
反应式:
[0101][0102]
称取90gtbscl溶解于100ml四氢呋喃中待用,将57.0g骨化三醇粗品用570ml四氢呋喃在室温下溶解,投入至2.0l三口反应瓶,加入83.8g咪唑,控温30℃以下滴加待用的tbscl的thf溶液,滴毕,保温25
±
5℃反应2~3h,tlc监控反应,反应完全后,加入285ml纯化水,搅拌3~5min,静置3~5min,分层,有机层收集,水层用285ml乙酸乙酯搅拌萃取3~5min,静置3~5min,分层,水层舍去,有机层合并,用171g无水硫酸钠搅拌干燥,过滤,滤液40~50℃下减压浓缩至无液滴滴出,再浓缩10~20min,得到淡黄色油状物,用513ml正己烷于40℃下常压溶解待用。正己烷润柱子,湿法上样,用正己烷/乙酸乙酯=100/1洗脱前杂,完毕后(注:采用薄层展开,展开剂:正己烷/乙酸乙酯=10/1),洗脱剂改为正己烷/乙酸乙酯=1/1,洗脱主产物点,完毕后,将主点洗脱液于40~50℃减压浓缩干,得到70g骨化三醇tbs保护物。
[0103]
将70g骨化三醇tbs保护物用700ml四氢呋喃溶解转移至2.0l三口反应瓶,加入553ml 1m
·
tbaf-thf,氮气置换三次并保护,升温50~60℃反应3~4h,tlc监控反应,反应完全后,40~50℃减压浓缩至干,馏底物加入700ml乙酸乙酯和700ml纯化水搅拌5~10min,静置3~5min,分层,水层舍去,有机相内加入140g无水硫酸钠,搅拌干燥30
±
5min,过滤,滤液于40~50℃减压浓缩至干,用350ml乙酸乙酯于40~50℃常压下溶解馏底物待用。正己烷润柱子,湿法上样,用正己烷:乙酸乙酯=5:1洗脱杂质,而后改用正己烷/乙酸乙酯=1/1洗脱主产物(产物未出现时,洗脱剂可重复使用),期间需要通过薄层监控主产物后面的杂质点(展开剂:正己烷/乙酸乙酯=1/2),经磷钼酸加热显色,确认与杂质点无交叉的主点洗脱剂都收集,而后于40~50℃减压浓缩至干,馏底物用其重量3~5倍体积的乙酸乙酯于40℃下溶解,而后滴加3~5倍乙酸乙酯体积的正己烷,滴毕,升温至50℃维持0.5h,降温至0℃,析晶0.5h,过滤,滤饼后于30~40℃干燥2h,得到25.80g骨化三醇,纯度99.22%,如图5。
[0104]
本发明通过上述方法有效分离去除了杂质pza和杂质pzb,经柱层析纯化,重结晶得到纯度较高的骨化三醇。按照面积归一化法分别计算发酵上清液、骨化三醇粗品、骨化三醇纯品的有关物质含量,结果见表5。
[0105]
表5
[0106][0107]
以上结果表明,经纯化后的骨化三醇含量高达99.67%,并有效控制杂质pzb的含量在0.1%以下。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1