一种盐酸罗哌卡因中间体的制备方法与流程
1.本发明涉及盐酸罗哌卡因中间体制备技术领域,具体涉及一种盐酸罗哌卡因中间体的制备方法。
背景技术:
2.盐酸罗哌卡因是一种新型长效酰胺类局麻药,与其他类似药物相比具有持效时间长、可控性好及毒副作用低等优点,使其需求量越来越大。随着需求量的增多,国内的研究力量也逐渐增大。
3.盐酸罗哌卡因中间体是制备盐酸罗哌卡因的主要片段,其结构式如下:
[0004][0005]
在申请号为cn201710470848.4、主题名称为一种盐酸罗哌卡因的制备方法的申请文件中,制备该中间体时,在步骤一采用甲苯和氯仿混合体系做溶剂,引入了二类溶剂氯仿,同时不利于溶剂回收套用,生产成本较高且后续处理过程用到超声操作,在工业化生产中不利于放大生产。
[0006]
在申请号为cn201910029895.4、主题名称为一种盐酸罗哌卡因中间体的制备及纯化方法的申请文件中公开了盐酸罗哌卡因中间体的合成方法,该合成方法中,采用手性原料为起始物,省去了拆分操作,但手性原料价格贵,且后续反应产物的精制收率偏低从而使得成本较高,反应过程如下:
[0007][0008]
在申请号为cn201410263792.1、主题名称为一种盐酸罗哌卡因的制备方法的申请文件中,使用2-哌啶甲酸为起始原料,与氯化氢气体反应控制ph为2-3,该过程中易出现成盐不充分而导致后续杂质较多的问题,且在后续操作中用二氯甲烷萃取后用硫酸钠干燥,在工业化生产中操作不方便,不利于工业化生产;该申请文件中的反应过程如下;
[0009][0010]
可见,目前虽然有多种盐酸罗哌卡因中间体的制备途径,但是这些制备方法由于在后处理中难度较大,导致难以工业应用;且在合成途径中的起始原料价格偏高、溶剂体系毒性大等问题,导致这些制备方法在工业应用受限。因此,提供一种适于工业应用的盐酸罗哌卡因中间体的制备方法对盐酸罗哌卡因合成来说具有重要意义。
技术实现要素:
[0011]
针对现有技术的上述不足,本发明提供了一种盐酸罗哌卡因中间体的制备方法,为盐酸罗哌卡因中间体的工业化生产提供了一条操作简单、生产成本低、生产效率高的工艺路线;使用该路线制备盐酸罗哌卡因中间体,收率在75.2%-78.0%之间,纯度>93%。
[0012]
本发明的技术方案如下:
[0013]
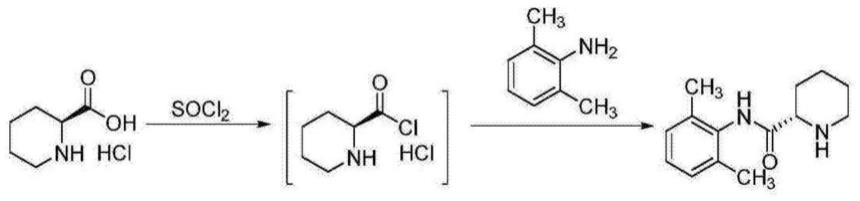
一种盐酸罗哌卡因中间体的制备方法,过程如下:
[0014]
步骤一:将2-哌啶甲酸加入到甲苯中,通入氯化氢气体进行成盐,通过控制终点ph确保成盐完全,得到2-哌啶甲酸盐酸盐-甲苯分散液;
[0015]
步骤二:向步骤一获得的2-哌啶甲酸盐酸盐-甲苯分散液中滴加碱催化剂,滴加过程控制溶液温度,滴加完毕后,搅拌均匀;然后继续在控温条件下滴加氯化亚砜,滴加完毕保温反应;
[0016]
步骤三:保温结束后,向溶液中控温滴加2,6-二甲基苯胺溶液,滴加完毕,保温;
[0017]
步骤四:步骤三保温结束后降温离心,得到固体湿品;将固体湿品用纯化水溶解后调节ph,然后使用有机溶剂萃取;将产品相调节ph,然后降温析晶,离心,烘干得到盐酸罗哌卡因中间体n-(2,6-二甲苯基)-2-哌啶甲酰胺。
[0018]
进一步的,在步骤一中,控制终点ph≤1.0,达到完全成盐。
[0019]
进一步的,在步骤一中,2-哌啶加酸与甲苯的重量比为1∶14-18。
[0020]
进一步的,在步骤二中,碱性催化剂为二乙胺、三乙胺、dmf或dmac中的一种。
[0021]
进一步的,在步骤二中,氯化亚砜的加入量根据2-哌啶甲酸的量确定,氯化亚砜与2-哌啶甲酸的重量比为1∶1-1.5。
[0022]
进一步的,步骤二和步骤三的保温温度均为50-70℃。
[0023]
进一步的,在步骤三中,2,6-二甲基苯胺与2-哌啶甲酸盐酸盐重量比为1.5∶1-2.0∶1。
[0024]
进一步的,在步骤四中,固体湿品溶解后,调节溶液的ph为5-6;萃取后,调节产品相的ph为10-12。
[0025]
进一步的,在步骤四中,萃取使用的有机溶剂为甲苯、乙酸乙酯或醋酸异丙酯中的一种。
[0026]
相对于现有技术,本发明的有益效果在于:
[0027]
1、本发明提供的盐酸罗哌卡因中间体合成方法,步骤一获得的2-哌啶甲酸盐酸不需要分离干燥即可直接进入下一步的反应中,有效降低操作难度,缩短操作时间,节省降温离心和烘干共计18小时每批次,提高生产效率。
[0028]
2、本发明提供的制备方法,通过将粗品溶解后,经萃取、析晶即可获得高纯度的盐酸罗哌卡因中间体,后处理过程简单、易于操作;另外,步骤四的萃取溶剂可蒸馏回收套用,使该制备方法适于工厂化应用。
[0029]
3、本发明提供的制备方法,为盐酸罗哌卡因中间体的工业化生产提供了一条操作简单、生产成本低、生产效率高的工艺路线;使用该路线制备盐酸罗哌卡因中间体,收率在75.2%-78.0%之间,纯度>93%。
附图说明
[0030]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,对于本领域普通技术人员而言,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0031]
图1为本发明盐酸罗哌卡因中间体的反应流程图。
[0032]
图2为实施例1制备的盐酸罗哌卡因中间体的纯度检测图谱。
[0033]
图3为实施例2制备的盐酸罗哌卡因中间体的纯度检测图谱。
[0034]
图4为实施例3制备的盐酸罗哌卡因中间体的纯度检测图谱。
[0035]
图5为实施例4制备的盐酸罗哌卡因中间体的纯度检测图谱。
[0036]
图6为实施例5制备的盐酸罗哌卡因中间体的纯度检测图谱。
具体实施方式
[0037]
为了使本技术领域的人员更好地理解本发明中的技术方案,下面将结合本发明的实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
[0038]
本发明以下的实施例中,二甲基苯胺溶液由2,6-二甲基苯胺与甲苯按照体积比为1∶1的比例组成。
[0039]
本发明中,色谱条件为:
[0040]
仪器型号:agilent 1260
[0041]
色谱柱信息:十八烷基硅烷键合硅胶为填充剂
[0042]
流动相:乙腈-磷酸盐缓冲液,(70∶30)
[0043]
检测波长:210nm;
[0044]
进样量:20μl;
[0045]
供试品溶液制备:
[0046]
称取本品20mg,加流动相溶解并稀释制成每1ml含0.2mg的溶液作为供试品溶液。
[0047]
实施例1
[0048]
结合图1的流程图,本发明提供了一种盐酸罗哌卡因中间体的制备方法,过程如
下:
[0049]
步骤一:
[0050]
向装有机械搅拌的500ml洁净四口瓶中加入2-哌啶甲酸17g和甲苯270g,室温下通入氯化氢气体;检测反应液的ph,当反应液的ph≤1时,停止通入氯化氢气体;得到2-哌啶甲酸盐酸盐-甲苯分散液;
[0051]
步骤二:向步骤一获得的2-哌啶甲酸盐酸盐-甲苯分散液中滴加2g dmf,滴加过程控制溶液温度在68-70℃之间,滴加完毕后,搅拌5分钟后,继续在68-70℃条件下滴加氯化亚砜25g,滴加完毕后在68-70℃条件下保温反应6小时,将剩余的氯化亚砜抽走;
[0052]
步骤三:步骤二保温结束后,继续在68-70℃条件下向反应液中滴加2,6-二甲基苯胺溶液65g,滴加完毕,在68-70℃条件下保温8小时;
[0053]
步骤四:在步骤三保温结束后,降低溶液温度至10-15℃,析出固体,离心,得到固体湿品31g,即为粗品,折干收率85%;
[0054]
步骤五,精制,将固体湿品用纯化水溶解后调节溶液的ph为5.5,然后使用乙酸乙酯萃取;分层,调节产品相的ph至10.8开始有固体析出,控制体系温度在10-15℃条件下保温2小时,过滤,滤饼干燥,得到盐酸罗哌卡因中间体23.8g,计算收率为78%,结合图2可知,液相纯度99.8%。
[0055]
实施例2
[0056]
一种盐酸罗哌卡因中间体的制备方法,过程如下:
[0057]
步骤一:
[0058]
向装有机械搅拌的500ml洁净四口瓶中加入2-哌啶甲酸17g和甲苯240g,室温下通入氯化氢气体;检测反应液的ph,当反应液的ph≤1时,停止通入氯化氢气体;得到2-哌啶甲酸盐酸盐-甲苯分散液;
[0059]
步骤二:向步骤一获得的2-哌啶甲酸盐酸盐-甲苯分散液中滴加2g dmac,滴加过程控制溶液温度在60-63℃之间,滴加完毕后,搅拌10分钟后,继续在60-63℃条件下滴加氯化亚砜20g,滴加完毕后在60-63℃条件下保温反应8小时,将剩余的氯化亚砜抽走;
[0060]
步骤三:步骤二保温结束后,继续在60-63℃条件下向反应液中滴加2,6-二甲基苯胺溶液60g,滴加完毕,在60-63℃条件下保温10小时;
[0061]
步骤四:在步骤三保温结束后,降低溶液温度至15-20℃,析出固体,离心,得到固体湿品27.2g,即为粗品,折干收率78.3%;
[0062]
步骤五,精制,将固体湿品用纯化水溶解后调节溶液的ph为5.1,然后使用甲苯萃取;分层,调节产品相的ph至11.6开始有固体析出,控制体系温度在15-20℃条件下保温2小时,过滤,滤饼干燥,得到盐酸罗哌卡因中间体23g,计算收率为75.2%,液相纯度99.8%,见图3。
[0063]
实施例3
[0064]
一种盐酸罗哌卡因中间体的制备方法,过程如下:
[0065]
步骤一:
[0066]
向装有机械搅拌的500ml洁净四口瓶中加入2-哌啶甲酸17g和甲苯300g,室温下通入氯化氢气体;检测反应液的ph,当反应液的ph≤1时,停止通入氯化氢气体;得到2-哌啶甲酸盐酸盐-甲苯分散液;
[0067]
步骤二:向步骤一获得的2-哌啶甲酸盐酸盐-甲苯分散液中滴加2g三乙胺,滴加过程控制溶液温度在64-67℃之间,滴加完毕后,搅拌20分钟后,继续在64-67℃条件下滴加氯化亚砜18g,滴加完毕后在64-67℃条件下保温反应8小时,将剩余的氯化亚砜抽走;
[0068]
步骤三:步骤二保温结束后,继续在64-67℃条件下向反应液中滴加2,6-二甲基苯胺溶液65g,滴加完毕,在64-67℃条件下保温6小时;
[0069]
步骤四:在步骤三保温结束后,降低溶液温度至15-20℃,析出固体,离心,得到固体湿品28.3g,即为粗品,折干收率80%;
[0070]
步骤五,精制,将固体湿品用纯化水溶解后调节溶液的ph为6.0,然后使用醋酸异丙酯萃取;分层,调节产品相的ph至10.5开始有固体析出,控制体系温度在10-20℃条件下保温3小时,过滤,滤饼干燥,得到盐酸罗哌卡因中间体23.4g,计算收率为76.5%,液相纯度99.8%,见图4。
[0071]
实施例4
[0072]
本发明提供了一种盐酸罗哌卡因中间体的制备方法,过程如下:
[0073]
步骤一:
[0074]
向5001搪玻璃釜中加入2-哌啶甲酸17kg和甲苯265kg,室温下通入氯化氢气体;检测反应液的ph,当反应液的ph≤1时,停止通入氯化氢气体;得到2-哌啶甲酸盐酸盐-甲苯分散液;
[0075]
步骤二:向步骤一获得的2-哌啶甲酸盐酸盐-甲苯分散液中滴加1.2kg dmf,滴加过程控制溶液温度在68-70℃之间,滴加完毕后,搅拌5分钟后,继续在68-70℃条件下滴加氯化亚砜18kg,滴加完毕后在68-70℃条件下保温反应6小时,将剩余的氯化亚砜抽走;
[0076]
步骤三:步骤二保温结束后,继续在68-70℃条件下向反应液中滴加2,6-二甲基苯胺溶液64kg,滴加完毕,在68-70℃条件下保温8小时;
[0077]
步骤四:在步骤三保温结束后,降低溶液温度至10-15℃,析出固体,离心,得到固体湿品33kg,即为粗品,折干收率83%;
[0078]
步骤五,精制,将固体湿品用纯化水溶解后调节溶液的ph为5.5,然后使用甲苯萃取;分层,调节产品相的ph至10.8开始有固体析出,控制体系温度在10-15℃条件下保温2小时,过滤,滤饼干燥,得到盐酸罗哌卡因中间体22.9kg,计算收率为75%,液相纯度99.8%,见图5。
[0079]
实施例5
[0080]
一种盐酸罗哌卡因中间体的制备方法,过程如下:
[0081]
步骤一:
[0082]
向装有机械搅拌的500ml洁净四口瓶中加入2-哌啶甲酸17g和甲苯300g,室温下通入氯化氢气体;检测反应液的ph,当反应液的ph≤1时,停止通入氯化氢气体;得到2-哌啶甲酸盐酸盐-甲苯分散液;
[0083]
步骤二:向步骤一获得的2-哌啶甲酸盐酸盐-甲苯分散液中滴加2g三乙胺,滴加过程控制溶液温度在64-67℃之间,滴加完毕后,搅拌20分钟后,继续在64-67℃条件下滴加氯化亚砜18g,滴加完毕后在64-67℃条件下保温反应8小时,将剩余的氯化亚砜抽走;
[0084]
步骤三:步骤二保温结束后,继续在64-67℃条件下向反应液中滴加2,6-二甲基苯胺溶液50g,滴加完毕,在64-67℃条件下保温6小时;
[0085]
步骤四:在步骤三保温结束后,降低溶液温度至15-20℃,析出固体,离心,得到固体湿品21.3g,即为粗品,折干收率62%;
[0086]
步骤五,精制,将固体湿品用纯化水溶解后调节溶液的ph为6.0,然后使用醋酸异丙酯萃取;分层,调节产品相的ph至10.5开始有固体析出,控制体系温度在10-20℃条件下保温3小时,过滤,滤饼干燥,得到盐酸罗哌卡因中间体17.5g,计算收率为57.2%,液相纯度93.6%,见图6。
[0087]
尽管通过参考优选实施例的方式对本发明进行了详细描述,但本发明并不限于此。在不脱离本发明的精神和实质的前提下,本领域普通技术人员可以对本发明的实施例进行各种等效的修改或替换,而这些修改或替换都应在本发明的涵盖范围内/任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应所述以权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1