一类芳香硫醚的制备方法与流程
1.本发明属于有机化合物技术领域,尤其是指一类芳香硫醚的制备方法。
背景技术:
2.在有机合成领域,硫醚是一类重要的有机硫化合物,是许多生物和医药活性分子合成的重要中间体。实际上,许多种类的芳香硫醚类化合物表现出了潜在的临床应用性质,尤其是具有药物活性的不对称芳香硫醚,其作为药物中间体已经被广泛应用于各种治疗领域,比如糖尿病、抗感染药、免疫、阿尔茨海默病、帕金森症等。
3.研究人员一直专注于开发各种构建碳硫键和过硫键的合成策略。近年来,随着社会对环境问题的关注,寻找更加绿色环保、可持续和高原子经济性的合成方法愈来愈受到科研人员的重视。因此开发新的高效率和通用性的策略来制备非对称硫醚显得极为重要。
技术实现要素:
4.为解决上述技术问题,本发明提供了一类芳香硫醚的制备方法。
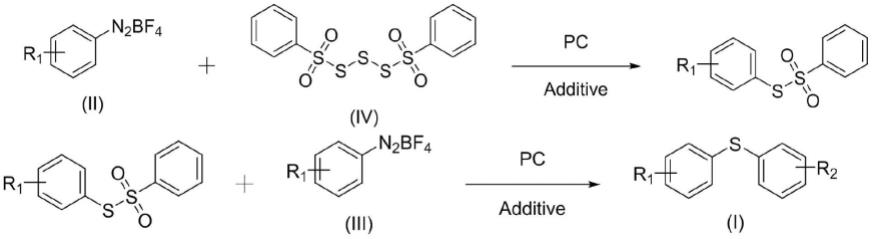
5.本发明的第一个目的在于提供一种芳香硫醚,所述芳香硫醚的结构为其中,r1、r2独立地选自烷基、取代或未取代的芳香基、取代或未取代的杂环基团。
6.在本发明的一个实施例中,所述取代的芳香基中的取代基团为烷基、卤素和烷氧基中的一种或多种。
7.在本发明的一个实施例中,所述杂环基团选自取代苯基、萘基或噻吩。
8.在本发明的一个实施例中,所述取代或未取代的杂环基团中取代基团为烷基、卤素和烷氧基中的一种或多种。
9.本发明的第二个目的在于提供所述的芳香硫醚的制备方法,包括以下步骤:在催化剂和添加剂的作用下,将与混合光照反应得到反应中间体之后之后混合进行光照反应,得到所述芳香硫醚。
10.在本发明的一个实施例中,所述催化剂选自三(2,2'-联吡啶基)氯化钌(ⅱ)六水合物、三(2-苯基吡啶)合铱、盐酸吖啶和2,4,5,6-四(9-咔唑基)-间苯二腈中的一种或多种。
11.在本发明的一个实施例中,所述添加剂选自磷酸钾、碳酸铯和磷酸氢钠中的一种
或多种。
12.在本发明的一个实施例中,所述反应的溶剂选自乙腈、乙醇和乙酸乙酯中的一种或多种。
13.在本发明的一个实施例中,所述反应的条件:室温下反应时间12-24h。
14.在本发明的一个实施例中,所述光照反应的光源选自波长为400nm-10nm的紫外光。
15.在本发明的一个实施例中,的摩尔比为1:1-1:2。
16.在本发明的一个实施例中,所述催化剂与的摩尔比为1:25-1:18。
17.在本发明的一个实施例中,所述添加剂与的摩尔比为1:25-1:18。
18.本发明的上述技术方案相比现有技术具有以下优点:
19.本发明所述的技术方案,以商业化的芳基重氮盐为原料,廉价易得,大大拓宽了芳基硫醚的底物范围,可以合成以往难以获得的芳基硫醚类化合物。
20.本发明所述的机理如下:
[0021][0022]
首先,在激发态光敏催化剂的作用下,重氮盐经历单电子转移过程形成芳基自由基并释放出氮气,芳基自由基与硫代磺酸酯反应生成芳基硫醚产物。
附图说明
[0023]
为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中
[0024]
图1是本发明实施例1化合物01的核磁氢谱谱图;
[0025]
图2是本发明实施例2化合物01的核磁氢谱谱图;
[0026]
图3是本发明实施例3化合物01的核磁氢谱谱图。
具体实施方式
[0027]
下面结合表1和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0028]
表1化合物结构及其1hnmr数据
[0029][0030]
实施例1
[0031]
本实例说明表1中化合物的具体合成方法。化合物可通过以下反应步骤合成:
[0032]
(1):中间体(a-1)与中间体(a-2)的合成
[0033][0034]
在50ml圆底烧瓶中,将杂环胺(10.6mmol)溶解于无水乙醇(8ml)和hbf4(50%,4ml,32mmol)混合溶液中,在0℃下缓慢滴加t-buono(6.5ml,48mmol)。不在析出重氮盐后,加入乙醚(4ml)。过滤混合物,用乙醚(10ml)和石油醚(10ml)洗涤,无需干燥即可获得所需的杂环重氮盐(a-1)与(a-2)。
[0035]
(2):中间体(a-3)与中间体(a-4)的合成
[0036][0037]
苯胺(10mmol)溶解于4ml蒸馏水和3.4ml 50%hbf4的混合物中,冷却至0℃后,在5min内缓慢滴加亚硝酸钠(0.69g加入1.5ml蒸馏水中),搅拌30min,过滤,收集沉淀,用丙酮重新溶解。加入乙醚直至四氟硼酸重氮盐沉淀,过滤,用3
×
10ml乙醚清洗三次,在真空下干燥,得到相应的中间体(a-3)与中间体(a-4)。
[0038]
(3):中间体(a-5)的合成
[0039][0040]
将苯亚磺酸钠(10g,61mmol)和硫(1.95g,61mmol)溶解在无水吡啶(60ml)中,得到黄色溶液。反应在氩气下搅拌,1小时后得到白色悬浮液。向悬浮液中加入乙醚,过滤反应物并用无水乙醚洗涤,得到phso2sna,为白色结晶固体。
[0041][0042]
将1.97g(10mmol)phso2sna溶解在三氯甲烷(10ml)中,缓慢加入0.5ml乙酰氯(7.2mmol),在室温下搅拌12小时。反应结束后,用乙酸乙酯和水萃取,除去水层,用无水硫酸钠干燥,旋蒸除去有机溶剂后,经柱层析提纯后得到淡黄色固体,即为相应的中间体(a-5)。
[0043]
(4):化合物的合成
[0044]
a)化合物01的合成
[0045][0046][0047]
手套箱中,称量0.2mmol化合物(a-1),0.4mmol化合物(a-5),ru(bpy)3cl2·
6h2o(5mol%)和k3po4(1eq.)于干燥的8ml反应小瓶中,加入2ml mecn溶剂,盖好反应瓶盖,室温及光照下反应12小时。反应结束后,在手套箱中称量0.2mmol化合物(a-4),盖好反应瓶盖,室温及光照下继续反应12小时,直至反应结束。旋蒸除去有机溶剂,经柱层析提纯后得到产物,即为化合物01,其1h nmr数据见表1。
[0048]
b)化合物02的合成
[0049]
[0050]
手套箱中,称量0.2mmol化合物(a-2),0.4mmol化合物(a-5),ru(bpy)3cl2·
6h2o(5mol%)和k3po4(1eq.)于干燥的8ml反应小瓶中,加入2ml mecn溶剂,盖好反应瓶盖,室温及光照下反应12小时。反应结束后,在手套箱中称量0.2mmol化合物(a-4),盖好反应瓶盖,室温及光照下继续反应12小时,直至反应结束。旋蒸除去有机溶剂,经柱层析提纯后得到产物,即为化合物02,其1h nmr数据见表1。
[0051]
c)化合物03的合成
[0052][0053][0054]
手套箱中,称量0.2mmol化合物(a-3),0.4mmol化合物(a-5),ru(bpy)3cl2·
6h2o(5mol%)和k3po4(1eq.)于干燥的8ml反应小瓶中,加入2ml mecn溶剂,盖好反应瓶盖,室温及光照下反应12小时。反应结束后,在手套箱中称量0.2mmol)化合物(a-4),盖好反应瓶盖,室温及光照下继续反应12小时,直至反应结束。旋蒸除去有机溶剂,经柱层析提纯后得到产物。即为化合物03,其1h nmr数据见表1。
[0055]
显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1