一种头孢氨苄钠的制备方法与流程
1.本发明属于药物合成技术领域,涉及一种兽用头孢类抗生素的制备方法,具体涉及一种头孢氨苄钠的制备方法。
背景技术:
2.头孢氨苄为广谱抗生素,对革兰氏阴性菌和革兰氏阳性菌均有抗菌作用,属于第一代头孢菌素类抗生素,于2014年我国批准为可兽用。头孢氨苄不易溶于水,目前市面上的兽用头孢氨苄制剂只有头孢氨苄片剂和注射用头孢氨苄混悬液。由于动物口服给药难度较大,并且向动物注射混悬液时,易引起动物的强烈痛感并且药物吸收率较差,因此头孢氨苄的这两种制剂形式均不利于动物给药。头孢氨苄钠为头孢氨苄的钠盐,易溶于水,制备头孢氨苄钠制剂粉针剂能够提高药物吸收效率,便于动物给药。目前国内市场上缺乏对头孢氨苄钠粉针剂的开发。
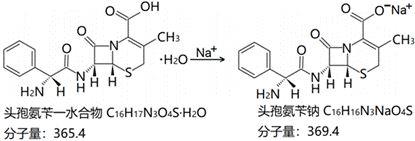
3.美国专利us3822256(a)公开了一种头孢氨苄钠的制备方法,具体步骤为:室温下,向头孢氨苄一水合物的异丙醇溶液中加入三乙胺和蒸馏水配制成澄清溶液;向上述澄清溶液中加入异丙醇,再加入钠盐溶液,搅拌反应;一定时间后,再次加入异丙醇,在0℃下搅拌反应;最后将反应混合物过滤、洗涤、干燥,得到白色晶体粉末。
4.虽然上述方法制备的头孢氨苄钠产率较高(≥89%),但头孢氨苄钠晶体形状为针状、棒状或球状-针状混合型,在工业化生产过程中,这些晶体形状的粉体流动性较差。
技术实现要素:
5.针对以上现有方法制得的头孢氨苄钠晶形较差、粉体流动性较差、不利于工业化生产的问题,本发明提供一种头孢氨苄钠的制备方法,该方法得到的头孢氨苄钠晶体粉末流动性好,利于工业生产。
6.为了实现上述目的,本发明采用以下技术方案:本发明提供一种头孢氨苄钠的制备方法,其特征在于,包括以下步骤:(1)向头孢氨苄溶液中加入供钠剂进行反应,或者,将所述头孢氨苄溶液和供钠剂同时加入底液进行反应;反应结束后,加入析晶溶剂养晶后得到待分离液;(2)对所述步骤(1)的待分离液依次进行过滤、洗涤、干燥,最终得到头孢氨苄钠;所述步骤(1)待分离液中包含丙酮和丙酮缩甘油,所述丙酮和丙酮缩甘油的总体积占待分离液体积的60~75%。
7.所述步骤(1)待分离液中的丙酮来自于头孢氨苄溶液和/或供钠剂和/或析晶溶剂;所述步骤(1)待分离液中的丙酮缩甘油来自于头孢氨苄溶液和/或供钠剂和/或析晶溶剂。
8.优选的,所述步骤(1)的头孢氨苄溶液中头孢氨苄的质量分数为8~11%。
9.优选的,所述步骤(1)头孢氨苄溶液的配制步骤包括:将头孢氨苄一水合物加入有机溶剂中,加入有机碱,搅拌使头孢氨苄一水合物溶解,得到澄清溶液。
10.进一步优选的,所述有机溶剂选自乙腈、丙酮、丙酮缩甘油、甲醇、乙醇、异丙醇、二氯甲烷、乙酸乙酯中的至少一种。
11.进一步优选的,所述头孢氨苄一水合物与所述有机溶剂的质量体积比为1:(10~15)g/m3。
12.有机碱能够将体系调至ph=8.0-10.0,促进头孢氨苄一水合物向头孢氨苄钠的转化。
13.进一步优选的,所述有机碱选自乙二胺、三乙胺、三乙醇胺、n,n-二甲基乙醇胺中的其中一种或几种。
14.进一步优选的,所述头孢氨苄一水合物溶解之后得到的溶液用活性炭进行吸附脱色处理并过滤。
15.优选的,所述头孢氨苄溶液的配制在25-35℃下进行;温度太低会影响溶解,温度太高会导致杂质偏高。
16.步骤(1)中反应式如式ⅰ所示:式ⅰ为控制反应速率,优化结晶状态,需要保证钠盐以更加分散的状态加入步骤(1)的反应体系内,因此,一般将钠盐加入分散剂中制成溶液或混悬液以提高分散性。
17.优选的,所述步骤(1)头孢氨苄溶液中头孢氨苄与供钠剂中钠元素的物质的量之比为1:(1.1-1.6)。
18.优选的,所述步骤(1)待分离液中丙酮与丙酮缩甘油的体积比为1:(1-2)。
19.优选的,所述步骤(1)中供钠剂为钠盐、所述钠盐与分散剂配成的钠盐溶液或钠盐混悬液中的任意一种。
20.进一步优选的,所述钠盐溶液或钠盐混悬液中钠盐的质量分数为15-30%。
21.进一步优选的,所述钠盐选自醋酸钠、乳酸钠、碳酸钠、异辛酸钠中的其中一种;所述分散剂选自乙腈、丙酮、丙酮缩甘油、碳酸二甲酯、甲乙酮、甲醇、乙醇、异丙醇、二氯甲烷、乙酸乙酯中的至少一种。
22.更进一步优选的,所述钠盐溶液的分散剂选自:乙腈、丙酮、丙酮缩甘油、甲醇、乙醇、甲乙酮、异丙醇;所述钠盐混悬液的分散剂选自:二氯甲烷、乙酸乙酯。
23.优选的,所述步骤(1)中底液为乙腈、丙酮、丙酮缩甘油、甲醇、乙醇、异丙醇、二氯甲烷、乙酸乙酯中的至少一种。
24.优选的,所述步骤(1)的反应温度为25-35℃,反应时间为1~2h。
25.优选的,所述步骤(1)析晶溶剂与待分离液的体积比为(0.4~0.7):1。
26.优选的,所述步骤(1)析晶溶剂选自乙腈、丙酮、丙酮缩甘油、碳酸二甲酯、甲乙酮、甲醇、乙醇、异丙醇、二氯甲烷、乙酸乙酯中的至少一种。
27.优选的,所述步骤(1)加入析晶溶剂后缓慢养晶1~2h得到待分离液。
28.步骤(2)中将所述头孢氨苄溶液和供钠剂同时加入底液的方法得到的产物晶形更加优异。
29.优选的,所述步骤(2)中洗涤采用的洗涤剂选自乙腈、丙酮、丙酮缩甘油、碳酸二甲酯、甲乙酮、甲醇、乙醇、异丙醇、二氯甲烷、乙酸乙酯中的至少一种。
30.优选的,所述步骤(2)干燥为在30~50℃下进行真空干燥。
31.步骤(1)中头孢氨苄溶液的有机溶剂、供钠剂的分散剂、底液和所述析晶溶剂与步骤(2)中的洗涤剂,均选自同一种溶剂,以便于母液回收处理。
32.一般地,药物晶体的尺寸和形状影响产品的质量及生产效率,而药物晶体的尺寸和形状易受生产工艺的影响,生产工艺中反应过程和析晶过程的溶剂是控制晶体尺寸及形状的重要因素之一。
33.依据本发明的方法制备的头孢氨苄钠晶体形状主要为不规则多面体形,该晶体形状的药物粉末通常过滤速度快、洗涤彻底、干燥时间短、流动性好,易实现大生产密闭转料等操作,这是因为:不规则多面体一般包含十几至几十个面,其不规则性导致颗粒间接触面较少,受力不平衡,从而导致颗粒间粘附力的减弱,在重力作用下其有更大的分散角度,即流动性。
34.本发明提供的一个或多个技术方案,至少具有以下技术效果:1、本发明的制备方法在析晶体系中包含一定比例的丙酮和丙酮缩甘油,利于不规则多面体形的头孢氨苄钠晶体的生长,得到的头孢氨苄钠粉末晶体在密闭转料过程中流动性显著提高。
35.2、本发明的制备方法中采用不同溶剂在反应体系中相互配合的方式,得到了高收率、高纯度的头孢氨苄钠。
36.3、本发明的制备方法反应条件温和,后处理操作简单,产品在密闭转料过程中流动性较高,利于车间生产。
附图说明
37.图1为实施例1中头孢氨苄钠晶体形状图片;图2为实施例1中头孢氨苄钠的dsc/tg检测结果;图3为实施例1中头孢氨苄钠的xrd检测结果;图4为实施例2中头孢氨苄钠的晶体形状图片;图5为实施例3中头孢氨苄钠的晶体形状图片;图6为实施例4中头孢氨苄钠的晶体形状图片;图7为实施例5中头孢氨苄钠的晶体形状图片;图8为实施例6中头孢氨苄钠的晶体形状图片;图9为对比例1中头孢氨苄钠的晶体形状图片;图10为对比例2中头孢氨苄钠的晶体形状图片;图11为对比例3中头孢氨苄钠的晶体形状图片;图12为对比例4中头孢氨苄钠的晶体形状图片;图13为对比例5中头孢氨苄钠的晶体形状图片;
图14为对比例6中头孢氨苄钠的晶体形状图片;图15为对比例7中头孢氨苄钠的晶体形状图片;图16为对比例8中头孢氨苄钠的晶体形状图片;图17为对比例9中头孢氨苄钠的晶体形状图片;图18为对比例10中头孢氨苄钠的晶体形状图片;图19为对比例11中头孢氨苄钠的晶体形状图片。
具体实施方式
38.以下配合说明书附图以具体实施例的方式对一种头孢氨苄钠的制备方法作进一步说明。
39.所有实施例及对比例中均采用梅特勒在线颗粒成像分析仪easyviewer 100 system对产物的晶形进行测定。
40.水分:药品中的水分包括结晶水和吸附水。水分含量的多少,对其稳定性、理化性状及药效作用等均有影响。药物粉针剂中水分的标准:4~6%。
41.药物粉针剂的ph标准:8.5~10.0。
42.总溶媒残留:即在原料药或赋形剂的生产中,以及在制剂制备过程中使用的有机挥发性化合物的残留;本发明中总溶媒残留通过高效气相色谱进行检测。
43.实施例1一种头孢氨苄钠的制备方法,包括以下步骤:(1)向250ml三口瓶中加入体积比为1:1的丙酮、丙酮缩甘油的混合液50ml,投入头孢氨苄一水合物5g(0.0137mol),控温25℃,滴加三乙醇胺搅拌至溶液澄清,加入0.5g活性炭,搅拌脱色30min,过滤得到质量分数为9%的头孢氨苄溶液;控温25~35℃,向头孢氨苄溶液中加入乳酸钠溶液(2.3g乳酸钠 0.0205mol 溶于10ml体积比为1:1的丙酮、丙酮缩甘油混合液中得到),搅拌20min,滴加体积比为1:1的丙酮、丙酮缩甘油的混合液100ml,缓慢搅拌养晶1小时,得到待分离液,待分离液中丙酮与丙酮缩甘油的总体积占待分离液体积的70%,待分离液中丙酮与丙酮缩甘油的体积比为1:1;(2)待分离液抽滤后得到晶体,用50ml体积比为1:1的丙酮、丙酮缩甘油的混合液洗涤晶体,真空40℃干燥2小时,得到头孢氨苄钠4.62g(0.0125mol),收率91.2%。
44.对产物进行测试分析,结果如表1中所示,晶体形状如说明书附图1所示,为不规则多面体形状,该形状的药物粉末在密闭转料过程中流动性很好,利于车间生产;对产物进行dsc/tg分析,结果如说明书附图2所示,产物不含结晶水,在270℃开始分解;对产物进行xrd分析,结果如说明书附图3所示,在2θ=5.73,8.57,9.00,9.40,11.45,15.01,17.31,18.52,19.22,20.57,22.20,22.88,23.96,25.21,27.39,28.64,29.51,31.03,32.94,36.73,38.90处有衍射峰,表明产物具有晶体形状。
45.实施例2一种头孢氨苄钠的制备方法,包括以下步骤:(1)向250ml三口瓶中加入体积比为1:1的丙酮与丙酮缩甘油的混合液50ml,投入头孢氨苄一水合物5g(0.0137mmol),控温25℃,滴加三乙胺搅拌至溶液澄清,加入0.5g活性炭,搅拌脱色30min,过滤得到质量分数为9%的头孢氨苄溶液;控温25~35℃,向50ml乙醇(底
液)中同时加入上述头孢氨苄溶液和醋酸钠溶液(1.68g醋酸钠0.0205mol 溶于10ml体积比为1:1的丙酮、丙酮缩甘油混合液),加完搅拌20min,滴加异丙醇100ml,缓慢搅拌养晶1小时,得到待分离液,待分离液中丙酮与丙酮缩甘油的总体积占待分离液体积的70%,待分离液中丙酮与丙酮缩甘油的体积比为1:1;(2)待分离液抽滤得到晶体,用50ml丙酮与丙酮缩甘油体积比为1:1的混合液洗涤晶体,真空40℃干燥2小时,得到头孢氨苄钠4.45g(0.012mmol),收率87.6%。
46.对产物进行测试分析,结果如表1中所示,晶体形状如说明书附图4所示,为碎颗粒状,在密闭转料过程中流动性较好。碎颗粒状产物在过滤时容易堵塞过滤介质微孔,从而导致过滤效果差,但因其为颗粒状,颗粒之间接触面少,受力不平衡,导致粘附力减弱,在外力影响下流动性较好。
47.实施例3一种头孢氨苄钠的制备方法,包括以下步骤:(1)向250ml三口瓶中加入丙酮50ml,投入头孢氨苄一水合物5g(0.0137mol),控温25℃,滴加三乙胺搅拌至溶液澄清,加入0.5g活性炭,搅拌脱色30min,过滤得到质量分数为10%的头孢氨苄溶液;控温25~35℃,向头孢氨苄溶液中加入乳酸钠的丙酮溶液(2.3g乳酸钠 0.0205mol 溶于10ml丙酮),加完搅拌20min,滴加体积比为1:3的丙酮、丙酮缩甘油混合液120ml,缓慢搅拌养晶1小时,得到待分离液,待分离液中丙酮体积占待分离液体积的70%;待分离液中丙酮与丙酮缩甘油的体积比为1:1;(2)待分离液抽滤后得到晶体,用50ml丙酮洗涤晶体,真空40℃干燥2小时,得到头孢氨苄钠4.55g (0.0123mmol),收率89.8%。
48.对产物进行测试分析,结果如表1中所示,晶体形状如说明书附图5所示,为不规则多面体形状,药物粉末在密闭转料过程中流动性较好。
49.实施例4一种头孢氨苄钠的制备方法,包括以下步骤:(1)向500ml三口瓶中加入丙酮缩甘油100ml,投入头孢氨苄一水合物10g(0.0274mol),控温25℃,滴加乙二胺搅拌至溶液澄清,加入1g活性炭,搅拌脱色30min,过滤得到质量分数为8%的头孢氨苄溶液;控温25~35℃,向头孢氨苄溶液中加入异辛酸钠的丙酮溶液(5.4g异辛酸钠 0.0325mol 溶于20ml丙酮),加完搅拌20min,滴加丙酮、丙酮缩甘油(体积比为2:1)混合液180ml,缓慢搅拌养晶1小时,得到待分离液,待分离液中丙酮与丙酮缩甘油的总体积占待分离液体积的70%,待分离液中丙酮与丙酮缩甘油的体积比为1:1.14;(2)待分离液抽滤得到晶体,100ml丙酮洗涤,真空40℃干燥2小时,得到头孢氨苄钠9.03g(0.0244mmol),收率89.1%。
50.对产物进行测试分析,结果如表1中所示,晶体形状如说明书附图6所示,为不规则多面体形状,药物粉末在密闭转料过程中流动性较好。
51.实施例5一种头孢氨苄钠的制备方法,包括以下步骤:(1)向250ml三口瓶中加入体积比为2:1的丙酮、丙酮缩甘油混合液60ml,投入头孢氨苄一水合物5g(0.0137mol),控温25℃,滴加三乙胺搅拌至溶液澄清,加入0.5g活性炭,搅拌脱色30min,过滤得到质量分数为8%的头孢氨苄溶液;控温25~35℃,向头孢氨苄溶液中加
入乳酸钠的丙酮溶液(2.3g乳酸钠 0.0205mol 溶于10ml丙酮),加完搅拌20min,滴加丙酮缩甘油60ml,缓慢搅拌养晶1小时,得到待分离液,待分离液中丙酮与丙酮缩甘油的总体积占待分离液体积的75%;(2)待分离液(待分离液中丙酮与丙酮缩甘油的体积比为1:1.6)抽滤得到晶体,50ml丙酮洗涤晶体,真空40℃干燥2小时,得到头孢氨苄钠4.58g(0.0124mmol),收率90.5%。
52.对产物进行测试分析,结果如表1中所示,晶体形状如说明书附图7所示,为不规则多面体形状,药物粉末在密闭转料过程中流动性较好。
53.实施例6一种头孢氨苄钠的制备方法,包括以下步骤:与实施例1相比,不同之处在于,步骤(1)至步骤(2)的丙酮、丙酮缩甘油的混合液中丙酮与丙酮缩甘油的体积比均为1:2,得到的头孢氨苄钠为4.58g(0.0124mol),收率为90.5%。
54.对产物进行测试分析,分析结果如表1中所示,晶体形状如说明书附图8所示,为不规则多面体形状,药物粉末在密闭转料过程中流动性较好。
55.对比例1一种头孢氨苄钠的制备方法,包括以下步骤:(1)向250ml三口瓶中加入丙酮50ml,投入头孢氨苄一水合物5g(0.0137mol),控温25℃,滴加乙二胺搅拌至溶液澄清,加入0.5g活性炭,搅拌脱色30min,过滤得到头孢氨苄溶液;控温25~35℃,向头孢氨苄溶液中加入醋酸钠的甲醇溶液(1.68g 0.0205mol 醋酸钠溶于5ml甲醇),加完搅拌20min,滴加丙酮100ml,缓慢搅拌养晶1小时,得到待分离液,待分离液中不包含丙酮缩甘油;(2)待分离液抽滤得到晶体,50ml丙酮洗涤,真空40℃干燥2小时,得到头孢氨苄钠4.3g(0.0116mol),收率84.7%。
56.对产物进行测试分析,分析结果如表1中所示,晶体形状如说明书附图9所示,为棒状,这种形状的晶体粉末在密闭转料过程中的流动性较差。
57.对比例2一种头孢氨苄钠的制备方法,包括以下步骤:(1)向250ml三口瓶中加入甲乙酮50ml,投入头孢氨苄一水合物5g(0.0137mol),控温25℃,滴加三乙胺搅拌至溶液澄清,加入0.5g活性炭,搅拌脱色30min,过滤得到头孢氨苄溶液;控温25~35℃,向头孢氨苄溶液中加入醋酸钠的甲乙酮溶液(1.68g 0.0205mol 醋酸钠溶于5ml甲乙酮),加完搅拌20min,滴加甲乙酮100ml,缓慢搅拌养晶1小时,得到待分离液,待分离液中不包含丙酮和丙酮缩甘油;(2)待分离液抽滤得到晶体,50ml甲乙酮洗涤晶体,真空40℃干燥2小时,得到头孢氨苄钠4.5g(0.0122mol),收率89.1%。
58.对产物进行测试分析,分析结果如表1中所示,晶体形状如说明书附图10所示,为针状,这种形状的晶体粉末在密闭转料过程中流动性很差。
59.对比例3一种头孢氨苄钠的制备方法,包括以下步骤:(1)向500ml三口瓶中加入碳酸二甲酯50ml,丙酮50ml,投入头孢氨苄一水合物10g
(0.0274mol),控温25℃,滴加乙二胺搅拌至溶液澄清,加入1g活性炭,搅拌脱色30min,过滤得到头孢氨苄溶液;控温25~35℃,向头孢氨苄溶液中加入异辛酸钠的丙酮溶液(5.4g异辛酸钠0.0325mol 溶于10ml丙酮),加完搅拌20min,滴加丙酮与碳酸二甲酯的混合液100ml(各50ml),缓慢搅拌养晶1小时,得到待分离液,待分离液中不含有丙酮缩甘油;(2)待分离液抽滤得到晶体,50ml乙醇洗涤晶体,真空40℃干燥2小时,得到头孢氨苄钠8.9g(0.0241mol),收率88.0%。
60.对产物进行测试分析,分析结果如表1中所示,晶体形状如说明书附图11所示,为棒状,这种形状的晶体粉末在密闭转料过程中的流动性较差。
61.对比例4一种头孢氨苄钠的制备方法,包括以下步骤:(1)向250ml三口瓶中加入异丙醇50ml,投入头孢氨苄一水合物5g(0.0137mol),控温25℃,滴加n,n-二甲基乙醇胺搅拌至溶液澄清,加入0.5g活性炭,搅拌脱色30min,过滤得到头孢氨苄溶液;控温25~35℃,向头孢氨苄溶液中加入异辛酸钠的异丙醇溶液(2.7g异辛酸钠 0.0162mol 溶于10ml异丙醇),加完搅拌20min,滴加异丙醇100ml,缓慢搅拌养晶1小时,得到待分离液,待分离液中不包含丙酮和丙酮缩甘油;(2)待分离液抽滤得到晶体,50ml异丙醇洗涤晶体,真空40℃干燥2小时,得到头孢氨苄钠4.6g(0.0125mol),收率91.2%。
62.对产物进行测试分析,分析结果如表1中所示,晶体形状如说明书附图12所示,为针状,这种形状的晶体粉末在密闭转料过程中的流动性较差。
63.对比例5一种头孢氨苄钠的制备方法,包括以下步骤:(1)向250ml三口瓶中加入乙醇50ml,投入头孢氨苄一水合物5g(0.0137mol),控温25℃,滴加三乙胺搅拌至溶液澄清,加入0.5g活性炭,搅拌脱色30min,过滤得到头孢氨苄溶液;控温25~35℃,向50ml乙醇(底液)中同时加入上述头孢氨苄溶液和醋酸钠的乙醇溶液(1.68g醋酸钠 0.0205mol 溶于5ml乙醇),加完搅拌20min,滴加异丙醇100ml,缓慢搅拌养晶1小时,得到待分离液,待分离液中不包含丙酮和丙酮缩甘油;(2)待分离液抽滤得到晶体,50ml异丙醇洗涤晶体,真空40℃干燥2小时,得到头孢氨苄钠4.3g(0.0116mol),收率84.7%。
64.对产物进行测试分析,分析结果如表1中所示,晶体形状如说明书附图13所示,为棒状,该种形状的晶体粉末在密闭转料过程中的流动性较差。
65.对比例6一种头孢氨苄钠的制备方法,包括以下步骤:与实施例1相比,不同之处在于,将步骤(1)至步骤(2)中的丙酮、丙酮缩甘油混合液全部替换为同等体积的乙醇,得到头孢氨苄钠4.30g(0.0116mol),收率为84.7%。
66.对产物进行检测分析,分析结果如表1中所示,晶体形状如说明书附图14所示,为棒状,这种形状的晶体粉末在密闭转料过程中的流动性较差。
67.对比例7与实施例1相比,不同之处在于,步骤(1)至步骤(2)的丙酮、丙酮缩甘油混合液中,丙酮和丙酮缩甘油的体积比均为1:0.5,得到的头孢氨苄钠为4.57g(0.0124mol),收率为
90.5%。
68.对产物进行测试分析,分析结果如表1中所示,晶体形状如说明书附图15所示,为棒状,这种形状的晶体粉末在密闭转料过程中的流动性较差。
69.对比例8与实施例1相比,不同之处在于,步骤(1)至步骤(2)的丙酮、丙酮缩甘油混合液中,丙酮和丙酮缩甘油的体积比均为1:4,得到的头孢氨苄钠为4.59g(0.0124mol),收率为90.5%。
70.对产物进行测试分析,分析结果如表1中所示,晶体形状如说明书附图16所示,为棒状,这种形状的晶体粉末在密闭转料过程中的流动性较差。
71.对比例9与实施例1相比,不同之处在于,步骤(1)至步骤(2)的丙酮、丙酮缩甘油混合液中,将丙酮替换为同等体积的异丙醇,得到的头孢氨苄钠为4.55g(0.0123mol),收率为89.8%,对产物进行测试分析,分析结果如表1中所示,晶体形状如说明书附图17所示,为棒状,这种形状的晶体粉末在密闭转料过程中的流动性较差。
72.对比例10与实施例1相比,不同之处在于,步骤(1)至步骤(2)的丙酮、丙酮缩甘油混合液中,将丙酮缩甘油替换为同等体积的异丙醇,得到的头孢氨苄钠为4.50g(0.0122mol),收率为89.1%。
73.对产物进行测试分析,分析结果如表1中所示,晶体形状如说明书附图18所示,为棒状,这种形状的晶体粉末在密闭转料过程中的流动性较差。
74.对比例11与实施例1相比,不同之处在于,将步骤(1)至步骤(2)的丙酮、丙酮缩甘油混合液均替换为异丙醇和乙醇的体积比为1:1的混合液,得到的头孢氨苄钠为4.45g(0.012mol),收率为87.6%。
75.对产物进行测试分析,分析结果如表1中所示,产物晶体形状如说明书附图19所示,为棒状,这种形状的晶体粉末在密闭转料过程中的流动性较差。
76.表1.实施例及对比例中头孢氨苄钠各项指标检测结果实施例或对比例水分ph钠含量旋光度总溶媒残留收率/%实施例14.5%9.65.7%142
°
0.45%91.2实施例23.9%8.75.5%141
°
0.35%87.6实施例34.6%9.35.1%141
°
0.43%89.8实施例44.1%9.55.5%141
°
0.27%89.1实施例54.3%9.45.6%143
°
0.38%90.5实施例64.8%9.15.2%143
°
0.40%90.5对比例14.6%9.35.1%141
°
0.43%84.7对比例24.1%9.55.5%141
°
0.27%89.1对比例34.3%9.45.3%143
°
0.38%88.0对比例43.7%9.75.9%142
°
0.35%91.2对比例54.2%9.75.4%142
°
0.23%84.7
对比例64.5%8.95.5%142
°
0.27%84.7对比例74.6%9.15.4%142
°
0.28%90.5对比例85.0%9.25.2%142
°
0.25%90.5对比例93.9%8.65.4%142
°
0.45%89.8对比例104.3%9.15.1%141
°
0.33%89.1对比例114.5%9.25.3%142
°
0.38%87.6结果分析:对比例1-6、9-11与实施例1-6相比,待分离液中不含或不同时含有丙酮与丙酮缩甘油,则所得头孢氨苄钠的晶体形状为针状或棒状,晶体粉末在密闭转料过程中流动性较差。
77.对比例7、对比例8与实施例1-6相比,待分离液中丙酮与丙酮缩甘油的体积比太大(2:1)或太小(1:4)不利于多面体形状晶体的形成,所得头孢氨苄钠的晶体形状多为棒状,在密闭转料过程中流动性较差。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1