一种蓝光磷化铟量子点及其制备方法、电致发光器件和显示装置与流程
1.本发明属于纳米技术领域,尤其涉及一种蓝光磷化铟量子点及其制备方法、电致发光器件和显示装置。
背景技术:
2.量子点具有半峰宽窄、量子产率高等优点,在显示、照明等领域有着巨大的应用前景。与ii-vi族元素量子点,如硒化镉量子点、碲化镉量子点等相比,以磷化铟量子点为代表的iii-v族元素量子点不含镉等高毒性重金属元素,应用范围更广,正逐渐受到科研界和产业界的关注。
3.然而,现有技术中合成蓝光磷化铟量子点比较困难,所合成的量子点质量差,在应用于电致器件中时,器件的导电性差、效率低、亮度低,完全不能满足应用使用的需求。因此,优化磷化铟量子点的制备方法,特别是蓝光磷化铟量子点的制备方法,具有非常重要的意义。
技术实现要素:
4.有鉴于此,本发明的目的是提供一种蓝光磷化铟量子点及其制备方法,由该蓝光磷化铟量子点制备得到的电致发光器件及显示装置,该蓝光磷化铟量子点的波长纯正,在应用于电致发光器件中时,亮度高、外量子效率也高。
5.为达到上述目的,本发明采用的技术方案是:
6.本发明的第一个目的在于提供一种蓝光磷化铟量子点的制备方法,包括如下步骤:
7.s1、将磷化铟量子点的核和第一锌前体混合,形成第一混合溶液;
8.s2、在300-340℃下,向所述第一混合溶液中加入硫醇进行反应,形成含有磷化铟量子点中间产物的第二混合溶液;
9.s3、在240-340℃下,向所述第二混合溶液中加入阴离子前驱体进行反应,得到所述蓝光磷化铟量子点;
10.其中,所述阴离子前驱体的反应活性低于所述硫醇的反应活性。
11.具体的,所述磷化铟量子点的核和第一锌前体的摩尔比为1:(10-100);
12.优选地,所述第一锌前体和所述硫醇的摩尔比为1:(1-5)。
13.具体的,所述硫醇和所述阴离子前驱体的摩尔比为1:(2-10),其中不包含1:2,且所述s3步骤中的反应温度为280-340℃,其中不包含280℃。
14.具体的,所述硫醇和所述阴离子前驱体的摩尔比为1:(0-2),其中不包含1:0,和/或,所述s3步骤中反应温度为240-280℃。
15.具体的,所述第一锌前体在300℃以上不会分解成氧化锌;
16.优选地,所述第一锌前体为卤代锌或脂肪酸锌。
17.具体的,所述阴离子前驱体为单质硒和/或单质硫的配位或者非配位溶液。
18.具体的,所述磷化铟量子点的核的制备方法如下,使铟前体、磷前体、第二锌前体与有机溶剂混合,于110~160℃下反应,得到包含磷化铟量子点的核的溶液,经纯化得到所述磷化铟量子点的核,其中,所述磷前体的化学结构式为m—(o—c≡p)n,其中,m为金属元素,n为1、2或者3。
19.本发明的第二个目在于提供一种蓝光磷化铟量子点,采用如上所述的制备方法制备得到,所述蓝光磷化铟量子点的波长为450-480nm。
20.本发明的第三个目的在于提供一种电致发光器件,包括发光层,采用如上所述蓝光磷化铟量子点作为发光层,所述电致发光器件的外量子效率大于0.5%,最大亮度大于100nits。
21.本发明的第三个目的在于提供一种显示装置,包括如上所述的电致发光器件。
22.与现有技术相比,本发明至少具有如下优点:通过本技术的制备方法,在预定温度下,将磷化铟量子点的核和第一锌前体混合,加入硫醇进行反应,形成含有磷化铟量子点中间产物的第二混合溶液;再加入反应活性低于硫醇的阴离子前驱体继续反应,得到波长范围在450~480nm的蓝光磷化铟量子点,该蓝光磷化铟量子点的波长纯正,在应用于电致发光器件中时,亮度高(大于100nits)、外量子效率也高(最高达到1.8%),拓宽了磷化铟量子点的应用范围。
附图说明
23.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
24.附图1为实施例1的蓝光磷化铟量子点溶液的荧光发射光谱图;
25.附图2为实施例1的蓝光磷化铟量子点溶液制备成器件的荧光发射光谱图;
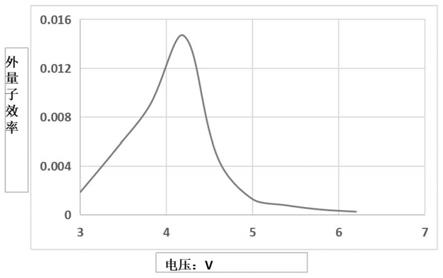
26.附图3为实施例1制备得到电致发光器件的电压-eqe变化图;
27.附图4为实施例1制备得到的电致发光器件的电压-亮度变化图;
28.附图5为实施例1的蓝光磷化铟量子点的透射电镜图。
具体实施方式
29.下面将结合本技术的实施方式,对实施例中的技术方案进行详细地描述。应注意的是,该实施方式仅仅是部分方式,而不是全部。
30.如本文中表述例如“的至少一种(个)”当在要素列表之前或之后时修饰整个要素列表而不修饰列表的单独要素。如果未另外定义,说明书中的所有术语(包括技术和科学术语)可如本领域技术人员通常理解的那样定义。常用字典中定义的术语应被解释为与它们在相关领域的背景和本公开内容中的含义一致,并且不可以理想方式或者过宽地解释,除非清楚地定义。此外,除非明确地相反描述,措辞“包括”和措辞“包含”当用于本说明书中时表明存在所陈述的特征、区域、整体、步骤、操作、要素、和/或组分,但是不排除存在或添加一个或多个其它特征、区域、整体、步骤、操作、要素、组分、和/或其集合。因此,以上措辞将
被理解为意味着包括所陈述的要素,但不排除任何其它要素。
31.如本文中使用的,术语“和/或”包括相关列举项目的一个或多个的任何和全部组合。术语“或”意味着“和/或”。
32.将理解,尽管术语第一、第二、第三等可在本文中用于描述各种元件、组分、区域、层和/或部分,但这些元件、组分、区域、层和/或部分不应受这些术语限制。
33.如本文中使用的“约”或“大约”包括所陈述的值且意味着在如由本领域普通技术人员考虑到所讨论的测量和与具体量的测量有关的误差(即,测量系统的限制)而确定的对于具体值的可接受的偏差范围内。例如,“约”可意味着相对于所陈述的值的偏差在一种或多种标准偏差范围内,或者在
±
10%、
±
5%范围内。
34.为了解决现有技术中,蓝光磷化铟量子点合成困难,现有技术中,磷化铟量子点波长调控难,尤其是蓝光磷化铟量子点的制备很困难,为了获得蓝光波长的磷化铟量子点,需要首要控制磷化铟量子点尺寸,但这也会带来应用于电致器件中,亮度低、外量子效率差的问题,不能满足应用要求的问题。
35.本发明提供了一种新的蓝光磷化铟量子点的制备方法,制备得到的蓝光磷化铟量子点的波长纯正,波长范围为450~480nm,半峰宽小于50nm,qy大于40%。
36.蓝光磷化铟量子点的制备方法具体包括如下步骤:
37.首先制备磷化铟量子点核:使铟前体(卤化铟)、磷前体(投料摩尔量大于铟前体的投料摩尔量)、第二锌前体(卤代锌)与配位溶剂混合,于110~160℃下反应,得到包含磷化铟量子点的核的溶液,经纯化得到磷化铟量子点的核,其中磷前体的化学结构式为m—(o—c≡p)n,其中,m为金属元素li、na、k、zn和ga中的一种,n为1、2或者3。上述纯化方法为现有技术中常用纯化方法,此处不作限定,只要能够达到纯化效果的方法即可。
38.接着,通过下述步骤继续反应:
39.s1、将磷化铟量子点的核和第一锌前体混合,形成第一混合溶液;
40.s2、在300-340℃下,向第一混合溶液中加入硫醇进行反应,形成含有磷化铟量子点中间产物的第二混合溶液;
41.s3、在240-340℃下,向第二混合溶液中加入阴离子前驱体进行反应,得到蓝光磷化铟量子点;阴离子前驱体的反应活性低于硫醇的反应活性。
42.本技术中,优选在300℃以上不会分解的第一锌前体,第一锌前体为卤代锌或脂肪酸锌;优选地,第一锌前体为选自氯化锌、硬脂酸锌、十一烯酸锌、十四酸锌、油酸锌中的至少一种。
43.阴离子前驱体为单质硒和/或单质硫的配位或者非配位溶液。磷化铟量子点的壳层为zns、znse和znses中的至少一种。发明人发现,将zns和/或znse和/或znses壳层生长在磷化铟量子点的表面,有利于获得具有更好稳定性和更优良电学性质的磷化铟量子点。
44.硫前体为硫的有机磷配合物、硫的脂肪胺化合物、硫的长链烯溶液中的至少一种;硒前体为硒的有机磷配合物、硒的脂肪胺化合物、硒的长链烯溶液中的至少一种。
45.本发明中,选择m—(o—c≡p)n作为合成磷化铟量子点的核的新磷源,制备得到磷化铟量子点核,金属元素m的引入,使得制备出具有由in、zn、p和金属元素m构成的磷化铟量子点核,进一步优化后续得到的磷化铟量子点的光学性能。
46.为了获得蓝光波长的磷化铟量子点,本发明选择磷化铟量子点的核和第一锌前体
的摩尔比为1:(10-100),第一锌前体和硫醇的摩尔比为1:(1-5)。通过上述原材料的摩尔比的选择,并在预设温度下反应,可以控制磷化铟量子点核的尺寸,获得蓝光波长的磷化铟量子点核。
47.将经纯化的磷化铟量子点核和第一锌前体混合,再在高温下与反应活性大的硫醇发生反应,可以获得含有波长纯正、荧光发射峰峰值范围在450~480nm的磷化铟量子点中间产物的第二混合溶液。本技术s2中,选择高温的反应条件和反应活性大的硫醇,是为了让zns壳层能够快速包裹到蓝光磷化铟的核上,防止蓝光磷化铟的核继续生长,从而获得波长纯正的磷化铟量子点的中间产物。
48.发明人发现,由于磷化铟量子点的中间产物的表面配体为活性高的硫醇,若直接应用于电致发光器件中,器件的导电性差,外量子效率低。因此,发明人通过向第二混合溶液中加入反应活性低于硫醇、位阻小于硫醇的阴离子前驱体,在预设温度下发生反应,以使让阴离子前驱体具有更高的反应能量,位阻小的阴离子前驱体使得量子点更易于导电,从而形成电致性能好的蓝光磷化铟量子点。
49.本发明的一个实施方式中,硫醇和阴离子前驱体的摩尔比为1:(2-10),其中不包含1:2,且s3步骤中反应温度为280-340℃,其中不包含280℃。通过本方法制备得到的蓝光磷化铟量子点应用于电致发光器件,器件的eqe达到1.1%以上。发明人认为,本实施方式中通过高温条件,使得硫醇与金属配位键断裂,再通过控制阴离子前驱体的用量远远大于硫醇的用量,使得阴离子前驱体能够与金属发生配位反应。具体有以下几种情况:
50.第一种:阴离子前驱体完全置换掉形成在磷化铟量子点核外与第一锌前体配位反应的硫醇,以在磷化铟量子点核外仅形成一层壳层,即阴离子前驱体与第一锌前体配位反应的第一壳层。
51.第二种:阴离子前驱体部分置换掉形成在磷化铟量子点核外与第一锌前体配位反应的硫醇,形成阴离子前驱体、硫醇分别与第一锌前体配位反应的第二壳层。第二壳层中的阴离子前驱体和第一锌前体的配位结合单元、硫醇和第一锌前体的配位结合单元是互相掺杂的关系。
52.第三种:阴离子前驱体部分置换掉形成在磷化铟量子点核外与第一锌前体配位反应的硫醇,形成阴离子前驱体、硫醇分别与第一锌前体配位反应的第二壳层。第二壳层中的阴离子前驱体和第一锌前体的配位结合单元、硫醇和第一锌前体的配位结合单元是互相掺杂的关系。此外,阴离子前驱体与多余的第一锌前体配位反应形成包裹在第二壳层外的第三壳层。
53.本发明的另一个实施方式中,硫醇和阴离子前驱体的摩尔比为1:(0-2),其中不包含1:0,和/或,s3步骤中反应温度为240-280℃。通过上述条件制备得到的蓝光磷化铟量子点,在应用于电致发光器件,器件的eqe达到0.5-1.1%,导电性远远优于现有技术中器件的eqe。
54.第四种:发明人认为,当阴离子前驱体的添加量不足和/或反应温度不足时,阴离子前驱体完全不能置换形成在磷化铟量子点核外与第一锌前体配位反应的硫醇。阴离子前驱体与多余的第一锌前体配位反应形成第四壳层,第四壳层包覆在硫醇与第一锌前体配位反应形成的壳层上。
55.因形成了第三壳层或第四壳层,需要多余的第一锌前体参与反应。优选地,s3中,
再次加入第一锌前体。本发明中,可以在s1步骤中一次性加入足够的第一锌前体,也可以分别在s1和s3步骤中分次加入第一锌前体。分次加入第一锌前体的方式,使得每步反应更加充分。
56.为了进一步提高所制备的蓝光磷化铟量子点光学性能,在得到上述蓝光磷化铟量子点后,还包括除去未反应的原料及其他杂质的步骤,具体包括分离和提纯。这些步骤是本领域的公知方法,这里不再赘述。
57.本发明的第三个目的在于提供一种电致发光器件,包括发光层,采用如上蓝光磷化铟量子点作为发光层,电致发光器件的外量子效率大于0.5%,最大亮度大于100nits。制备方法采用本领域公知方法即可,这里不再赘述。
58.本发明的第四个目的在于提供一种显示装置,包括如上电致发光器件。
59.以下将以具体的实施例对本技术做出详细的阐述。
60.实施例1、本实施例提供一种蓝光磷化铟量子点的制备方法,具体步骤如下:
61.制备磷化铟量子点核:将0.5mmol氯化铟、0.75mmol na—o—c≡p、1mmol的氯化锌与10ml油胺混合,于160℃下反应30min,得到磷化铟量子点核溶液,经纯化得到磷化铟量子点核,待用。
62.s1、配制40ml,浓度为0.5m的硬脂酸锌的十八烯溶液,120℃抽真空30分钟,转成氩气,升温到240℃,加入上述制备得到的磷化铟量子点核的十分之一中,形成第一混合溶液;
63.s2、在240℃下滴加十二硫醇进行反应,滴加速度为5ml/h,滴加1小时,同时继续升温到310℃,保温至十二硫醇滴加完毕,形成第二混合溶液;
64.s3、降温至290℃,向第二混合溶液中滴加top-s(浓度为2m)进行反应,速度为10ml/h,滴加2小时,降温,纯化,得到蓝光磷化铟量子点。将上述制备得到的蓝光磷化铟量子点制备成电致发光器件、显示装置,制备方法采用现有技术中常规方法,此处不赘述。
65.实施例2、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例1基本相同,不同之处在于,阴离子前驱体的添加量不同:s3中,添加top-s(浓度为2m),速度为10ml/h,滴加4.5小时。
66.实施例3、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例1基本相同,不同之处在于,阴离子前驱体的添加量不同:s3中,添加top-s(浓度为2m),速度为10ml/h,滴加9小时。
67.实施例4、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例1基本相同,不同之处在于,阴离子前驱体的添加量不同:s3中,添加top-s(浓度为2m),速度为10ml/h,滴加0.5小时。
68.实施例5、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例1基本相同,不同之处在于,阴离子前驱体的添加量不同:s3中,添加top-s(浓度为2m),速度为10ml/h,滴加1.8小时。
69.实施例6、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例1基本相同,不同之处在于,阴离子前驱体参与反应的温度不同:s3中,降温至240℃,再加入top-s。
70.实施例7、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例1基本相同,不同之处在于,阴离子前驱体的种类不同:s3中,加入的阴离子前驱体为top-se(浓度为2m)。
71.实施例8、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例1基本相同,不同之处在于,阴离子前驱体的种类不同:s3中,加入的阴离子前驱体为top-se、top-s(浓度为2m),速度各为5ml/h,滴加2小时。
72.实施例9、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例1基本相同,不同之处在于,增加十二硫醇的用量:s2中,十二硫醇的滴加速度为10ml/h,滴加1小时。
73.实施例10、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例1基本相同,不同之处在于,降低十二硫醇的用量:s2中,十二硫醇的滴加速度为3ml/h,滴加1小时。
74.实施例11、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例1基本相同,不同之处在于,将硫醇改变为短链硫醇:s2中,滴加的硫醇为正辛硫醇。
75.实施例12、本实施例提供一种蓝光磷化铟量子点的制备方法,其与实施例3基本相同,不同之处在于,分两次加入锌源,具体步骤如下:s1、配制20ml,浓度为0.5m的硬脂酸锌的十八烯溶液,s3中,降温至290℃,加入20ml的0.5m的硬脂酸锌的十八烯溶液,再向第二混合溶液中滴加top-s(浓度为2m)进行反应。
76.对比例1、本对比例提供一种磷化铟量子点的制备方法,具体步骤如下:配制40ml,浓度为0.5m的硬脂酸锌的十八烯溶液,120℃抽真空30分钟,转成氩气,升温到240℃,加入上述备用的核(制备方法同实施例1)十分之一,待温度重新升温到240℃后,开始滴加top-s(浓度为2m),滴加速度为5ml/h,滴加1小时,同时继续升温到310℃,保温至top-s滴加完毕。降温,纯化,得到磷化铟量子点。将上述制备得到的蓝光磷化铟量子点制备成电致发光器件、显示装置,制备方法采用现有技术中常规方法,此处不赘述。
77.对比例2、本对比例提供一种磷化铟量子点的制备方法,具体步骤如下:配制40ml,浓度为0.5m的硬脂酸锌的十八烯溶液,120℃抽真空30分钟,转成氩气,升温到240℃,加入上述备用的核(制备方法同实施例1)十分之一,待温度重新升温到240℃后,开始滴加十二硫醇,滴加速度为5ml/h,滴加1小时,同时继续升温到310℃,保温至十二硫醇滴加完毕。降温,纯化,得到磷化铟量子点。将上述制备得到的蓝光磷化铟量子点制备成电致发光器件、显示装置,制备方法采用现有技术中常规方法,此处不赘述。
78.将实施例1-12获得的蓝光磷化铟量子点和对比例1-2中获得的磷化铟量子点分别制备成电致发光器件,测试其荧光光谱和荧光量子产率。
79.具体测试结果如下表所示。
[0080][0081][0082]
从上述实施例及对比例可以看出,经本技术的方法制备得到的蓝光磷化铟量子点的波长纯正,从附图2中可看出,荧光发射峰峰值范围在450~480nm。在应用于电致发光器件中时,亮度高且大于100nits、外量子效率最高达到1.8%,拓宽了磷化铟量子点的应用范围。
[0083]
附图3为实施例1制备得到电致发光器件的电压-eqe变化图,能够看出最大eqe为1.5%。
[0084]
附图4为实施例1制备得到的电致发光器件的电压-亮度变化,可以看出最大亮度为124nits。
[0085]
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1