适用于井下压裂作业的可降解暂堵球及其制备方法与流程
1.本发明属于油气井开采技术领域,具体涉及一种适用于井下压裂作业的可降解暂堵球及其制备方法。
背景技术:
2.近年来,随着油气田的不断开采,储层的品质会逐年下降,为此相关的油气开采商多采用储层压裂改造的方式来改善地下储层的渗透性,以继续有效地从这些地下储层中开采油气资源。目前,在油气田的井下压裂作业中,通常需要使用暂堵球,而现有暂堵球一般是以橡胶、非可降解塑料、合金等材料制备而成,但由橡胶或非可降解塑料制成的暂堵球在实际应用时,往往难以溶解,容易被卡在炮眼中,难以在暂堵转向压裂施工结束后返排出井眼,为此必须采取措施对其进行钻磨或打捞,这不仅会大大增加作业成本,降低生产效率,并且在钻磨过程中通常会用到更多的液体,容易对地下储层造成二次污染。另外,由合金(例如,镁合金)制成的暂堵球,虽然刚性大且强度高,但其对溶解环境具有选择性,需要在较高的离子浓度下才能溶解,且溶解后具有残渣,容易对地下储层造成污染;当遇到炮眼形状不规则的情况时,由于合金材料的刚性大,其难以产生适应于不规则形状炮眼的形变,以致其对炮眼的密封性较差,难以形成稳定的承压强度,进而影响暂堵转向压裂的效果。
技术实现要素:
3.本发明的目的就是为了解决上述问题而提供一种适用于井下压裂作业的可降解暂堵球及其制备方法。
4.本发明的目的通过以下技术方案实现:
5.一种适用于井下压裂作业的可降解暂堵球,其是由包括以下组分及其重量份含量的原料制备而成:聚乙醇酸70
‑
90份、柔性可降解树脂10
‑
30份、相容剂4
‑
8份、增塑剂2
‑
5份、扩链剂0.5
‑
2份、功能填料15
‑
30份、抗氧剂0.2
‑
1.6份、热稳定剂2
‑
4份以及抗水解剂0.1
‑
1份。
6.作为一种实施方案,所述聚乙醇酸的分子量分布指数(m
w
/m
n
)为1.2
‑
1.5,且重均分子量m
w
为10
‑
20万。
7.作为一种实施方案,所述柔性可降解树脂选自聚丁二酸乙二醇酯、聚丁二酸丁二醇酯、聚己二酸
‑
对苯二甲酸
‑
丁二醇酯、聚丁二酸
‑
对苯二甲酸
‑
丁二醇酯、聚丁二酸
‑
己二酸
‑
丁二醇酯、聚甲基乙撑碳酸酯、或聚己二酸乙二醇酯中的至少一种。
8.作为一种实施方案,所述相容剂选自eba
‑
g
‑
mah或eva
‑
g
‑
mah中的至少一种。
9.优选地,所述eba
‑
g
‑
mah中ba含量为16
‑
18wt%,且熔融指数≤10g/10min(190℃,2.16kg)。
10.优选地,所述eva
‑
g
‑
mah中va含量为20
‑
28wt%,且熔融指数≤20g/10min(190℃,2.16kg)。
11.作为一种实施方案,所述增塑剂选自市售的甘油、环氧大豆油、环氧糠油酸丁酯或
乙酰柠檬酸三丁酯中的至少一种。
12.作为一种实施方案,所述扩链剂为环氧类扩链剂,例如,可选自basf生产的adr 4370s。
13.作为一种实施方案,所述功能填料为95%过800目筛的改性沸石粉。
14.所述改性沸石粉的制备方法如下:
15.1)将沸石粉加入至浓度为10
‑
25wt%的naoh溶液中,于50
‑
60℃的水浴中搅拌处理20
‑
60min,后经固液分离,保留固体,并用去离子水洗涤至中性,再置于80
‑
100℃的烘箱中干燥4
‑
8h,即制得预处理沸石粉;
16.2)将预处理沸石粉加入至含有γ
‑
氨丙基三甲氧基硅烷和3
‑
(2,3
‑
环氧丙氧)丙基甲基二甲氧基硅烷的乙醇溶液中,于80
‑
90℃的水浴中搅拌处理1
‑
2h,制得悬浮液,再将悬浮液转移至超声波分散机,于35
‑
60℃下超声处理1
‑
2h,后经固液分离,保留固体,采用无水乙醇洗涤数次,于80
‑
100℃的烘箱中干燥10
‑
24h,经研磨,即制得改性沸石粉。
17.优选地,上述步骤2)所采用的乙醇溶液是由无水乙醇和去离子水按质量比为1:1
‑
5混合而成。
18.优选地,上述步骤2)中,所述预处理沸石粉和乙醇溶液的用量关系为:每100g乙醇溶液中加入10
‑
25g预处理沸石粉。
19.优选地,上述步骤2)中,所述γ
‑
氨丙基三甲氧基硅烷的用量为预处理沸石粉质量的1.2
‑
1.8%,所述3
‑
(2,3
‑
环氧丙氧)丙基甲基二甲氧基硅烷的用量为预处理沸石粉质量的0.4
‑
0.8%。
20.优选地,上述步骤2)中,所述超声波分散机的超声处理功率为400
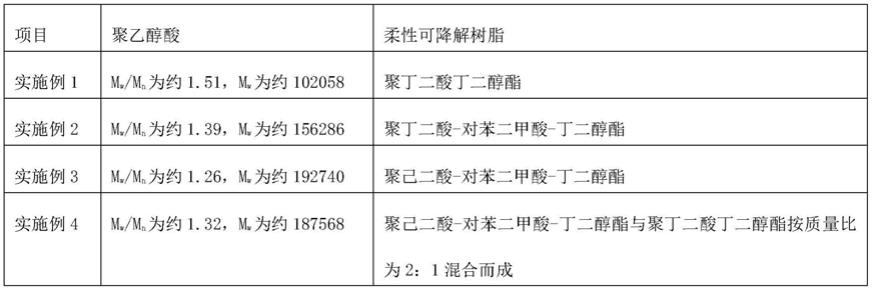
‑
600w。
21.优选地,所述固液分离可采用本领域常规的过滤、抽滤、离心分离等方法来实现,在此不再赘述。
22.作为一种实施方案,所述抗氧剂选自β
‑
(3,5二叔丁基
‑4‑
羟基苯基)丙酸十八醇酯、1,1,3三(2
‑
甲基
‑
4羟基
‑
5叔丁基苯基)丁烷、2,2
’‑
甲撑双(4
‑
乙基
‑
6叔丁基苯酚)、1,3,5
‑
三(3,5叔丁基
‑4‑
羟基苄基)三甲基苯、2,2
’‑
甲撑双(4
‑
甲基
‑6‑
叔丁基苯酚)或4,4
’‑
二叔辛基二苯胺中的至少一种。
23.作为一种实施方案,所述热稳定剂为有机锡类热稳定剂,优选为马来酸二丁基锡、二月桂酸二丁基锡、月桂酸马来酸二丁基锡、马来酸二正辛基锡、二月桂酸二正辛基锡或双(硫代甘醇酸异辛酯)二正辛基锡中的至少一种。
24.作为一种实施方案,所述抗水解剂为双(2,6
‑
二异丙基苯基)碳二亚胺。
25.上述可降解暂堵球的制备方法如下:
26.步骤1)熔融聚乙醇酸的制备:
27.步骤1
‑
1)将纯化的乙交酯粉末导入至熔融混合釜中,于常压下升温至110
‑
120℃,边搅拌边加入适量的反应助剂,使熔融的乙交酯与反应助剂混合均匀,获得呈流体态的预混料;
28.步骤1
‑
2)将呈流体态的预混料输送至静态混合器中进行预聚合,获得具有一定分子量的乙醇酸预聚物(例如,重均分子量为约5
‑
15万),随后将所述乙醇酸预聚物与交联剂一起输送至增粘设备中进行终聚合,以获得熔融聚乙醇酸;
29.步骤2)成型粒料的制备:按重量份将柔性可降解树脂、相容剂、增塑剂、扩链剂、功
能填料、抗氧剂、热稳定剂和抗水解剂加入至混合挤出设备中,先进行塑化,获得塑化预混料,然后将增粘设备制得的熔融聚乙醇酸直接导入至混合挤出设备的共混段,使其与塑化预混料进行共混,再由混合挤出设备的挤出段挤出、造粒,即制得成型粒料;
30.步骤3)球体成型:将成型粒料置于注塑机中,加热至熔融状态,注射到具有设定直径的球形模具的内腔中,将球形模具自然冷却以消除残余应力,即制得可降解暂堵球。
31.在此需要说明的是,本发明中“增粘设备”起脱挥作用,可促使乙醇酸预聚物的进一步聚合,及时将生成的小分子脱除体系,以使聚合物的分子量得到进一步提升,对应地聚合物粘度也会进一步增大。本发明中使用的增粘设备可以为,例如,仅设有脱挥段的双螺杆挤出机。本发明中使用的混合挤出设备可以为,例如,设有塑化段、共混段及挤出段的双螺杆挤出机。
32.在上述制备方法中:
33.优选地,所述纯化的乙交酯粉末的d
90
≤200μm,纯度≥98.5%,酸度≤20mmol/kg。
34.优选地,所述反应助剂包含催化剂、引发剂及脱水剂。
35.在反应助剂用量方面,所述催化剂的用量为乙交酯质量的约0.001
‑
1wt%,所述引发剂的用量为乙交酯质量的约0.1
‑
1wt%,所述脱水剂的用量为乙交酯质量的约0.2
‑
1.6wt%。
36.优选地,所述催化剂可选自锡类化合物、锑类化合物或锌类化合物中的至少一种,例如但不限于辛酸亚锡、氯化亚锡、乳酸锡、三氧化二锑、二乙基锌或二水合乙酸锌。
37.优选地,所述引发剂可选自具有伯醇或仲醇等羟基结构的烷烃类物质(例如,正丙醇、异丙醇、正丁醇、异丁醇等)或具有羟基活性基团的芳香类物质(例如,苯甲醇、苯乙醇等)中的一种或两种。
38.优选地,所述脱水剂可选自碳化二亚胺、聚碳化二亚胺或基于碳化二亚胺的化合物(例如但不限于n,n
’‑
二异丙基碳二亚胺、二环己基碳二亚胺等)。
39.为防止出现熔融乙交酯局部反应助剂浓度过高,可采用现有的注射方式将反应助剂逐滴加入至熔融混合釜中。
40.优选地,所述静态混合器中预聚合的温度控制为150
‑
230℃。
41.优选地,所述静态混合器可选自市售的sk型静态混合器。
42.优选地,所述静态混合器采用多段阶梯升温方式,例如,静态混合器共设有四段,第一段温度设定为150
‑
170℃,第二段温度设定为190
‑
210℃,第三段温度设定为210
‑
220℃,第四段温度设定为220
‑
230℃。
43.在实际制备过程中,物料在静态混合器中的总时长一般不超过150min,物料通过静态混合器的各段所需时间可以为:例如但不限于,通过第一段为约3
‑
8min,通过第二段为约10
‑
15min,通过第三段为约15
‑
20min,通过第四段为约50
‑
60min。
44.优选地,所述交联剂选自官能度≥3的多羟基烷烃类化合物(例如,双季戊四醇、丙三醇、三羟甲基乙烷、三羟甲基丙烷等)或含有不饱和烃的硅烷类化合物(例如,乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、乙烯基三(b
‑
甲氧基乙氧基)硅烷、γ
‑
(甲基丙烯酰氧)丙基三甲氧基硅烷等)中的至少一种。
45.在交联剂用量方面,所述交联剂的用量为基于乙交酯的质量计算所得聚乙醇酸理论质量的约1
‑
30wt%。
46.所述增粘设备仅设有脱挥段,该脱挥段的绝压≤200pa,且温度设定为220
‑
250℃。
47.优选地,所述脱挥段的螺杆长径比设定为50
‑
60。
48.在实际生产过程中,所述交联剂可由脱挥段的起始处加入。
49.所述混合挤出设备中沿物料的进给方向依次设有塑化段、共混段及挤出段。
50.优选地,所述增粘设备的出料口与混合挤出设备中共混段的起始处相连接。
51.优选地,所述混合挤出设备中塑化段的温度控制为130
‑
160℃,共混段的温度控制为200
‑
220℃,挤出段的温度控制为220
‑
230℃。
52.优选地,所述混合挤出设备的螺杆长径比为40
‑
50。
53.所述注塑机中,喂料温度控制为150
‑
160℃,计量段温度控制为190
‑
210℃,喷射口温度控制为230
‑
250℃,注塑压力控制为40
‑
90mpa,球形模具温度控制为120
‑
160℃。
54.与现有技术相比,本发明具有以下优点:
55.1、本发明暂堵球的材料体系是以聚乙醇酸为基体,引入柔性可降解树脂,通过相容剂来改善聚乙醇酸和柔性可降解树脂之间的相容性,使两者能够有效地复合在一起,在保证材料体系具有较高机械强度的同时,还可赋予材料体系一定的柔韧性,以使得制成的暂堵球在经受一定压力的作用下,可发生一定程度的自适应弹性形变,能够更好地封堵住形状不规则的射孔,从而发挥相对持久稳定的封堵承压作用,另外,为了进一步提高材料体系在高温条件下的承压稳定性,本发明将经硅烷偶联剂改性的沸石粉引入到材料体系中,经改性的沸石粉表面具有丰富的活性官能团,其可与聚乙醇酸、柔性可降解树脂之间形成强的作用,并在相容剂的调和作用下,能够均匀地分散在材料体系中,其可与抗氧剂、热稳定剂发挥协同增效作用,不仅可有效提高材料体系的耐热稳定性,使得制成的暂堵球在高温条件下依然能够维持相对较好的机械强度和韧性,从而保持相对稳定的封堵承压作用,而且由于沸石粉自身具有丰富的多孔结构,在暂堵球的后期自降解过程中,多孔结构又容易将外界液体(例如具有一定酸性的液体)引入至暂堵球的材料体系中,从而加速暂堵球的崩裂、降解,且无任何污染。
56.2、本发明通过熔融混合釜将乙交酯和反应助剂充分混合均匀以获得预混料,再将其导入至静态混合器中进行预聚合,静态混合器可对预混料起到低剪切高分散的作用,有利于防止并消除物料内局部区域的热量积累,有效防止物料内因受热不均而导致局部区域温度过高并引发热降解等副反应的发生,从而保证乙交酯能够进行良好的预聚合反应,获得具有一定分子量的乙醇酸预聚物,再将乙醇酸预聚物导入至增粘设备(例如,仅设有脱挥段的双螺杆挤出机)中进行终聚合,可有效缩短物料在增粘设备中受高剪切作用的时间,不仅有利于抑制热降解等副反应的发生,还有利于抑制酯交换反应的发生,从而减少体系中低聚物和/或低分子链物质的含量,制得的聚乙醇酸不仅分子量可得到显著提升,而且分子量分布指数相对较小,分子量分布相对均匀,这有利于改善材料的耐热稳定性。
57.3、本发明将熔融的乙交酯的流体态预混料导入至静态混合器中,利用交叉流动的方式以强化乙交酯和反应助剂之间的混合效果,使得反应助剂能够更加均匀地分散在反应体系中,同时利用梯度升温的方式,先在相对低的温度下和相对短的时间内较为温和地引发乙交酯的开环聚合反应,随后适当地升高温度并适当地延长时间,以在反应体系中形成较为稳定的且具有反应活性的乙醇酸分子链,再于相对高的温度下和相对长的时间内促使乙醇酸分子链的进一步增长,以获得具有一定分子量的乙醇酸预聚物,上述静态混合器的
梯度升温过程有利于抑制反应体系粘度的急剧变化,可有效避免由于反应体系局部粘度的急剧变化而造成热量过量积累以致结焦结渣现象的发生,另外,本发明在增粘设备脱挥段的起始处加入交联剂,通过交联剂向聚乙醇酸中引入适量的三维网状交联结构,在兼顾材料力学性能的同时,可有效改善材料的耐热稳定性。
58.4、本发明在成型粒料的制备过程中,由经静态混合器的预聚合、增粘设备的终聚合而获得的熔融的聚乙醇酸直接被导入至混合挤出设备的共混段,与改性组分在适应于聚乙醇酸加工温度的条件下进行共混改性,一方面可略去对聚乙醇酸物料进行二次熔融塑化的操作,不仅可缩短工序,提高生产效率,还可避免因聚乙醇酸物料的二次熔融而造成其自身热降解程度恶化的现象发生,另一方面改性组分(主要是指柔性可降解树脂)可在适应于其自身加工温度的条件下进行塑化,进而避免了其与聚乙醇酸在适应于聚乙醇酸加工温度的条件下进行熔融塑化而导致其自身严重热降解的现象发生。
59.5、本发明方法适用于工业放大生产,可利用现有的生产设备进行生产线改造,灵活适用性好,实现低碳化生产,具有很好的经济效益。
附图说明
60.图1为增粘设备与混合挤出设备的连接关系示意图;
61.图中:100
‑
增粘设备;200
‑
混合挤出设备;1
‑
脱挥段;2
‑
塑化段;3
‑
共混段;4
‑
挤出段。
具体实施方式
62.下面结合附图和具体实施例对本发明进行详细说明。
63.一种适用于井下压裂作业的可降解暂堵球,由包括以下组分及其重量份含量的原料制备而成:聚乙醇酸70
‑
90份、柔性可降解树脂10
‑
30份、相容剂4
‑
8份、增塑剂2
‑
5份、扩链剂0.5
‑
2份、功能填料15
‑
30份、抗氧剂0.2
‑
1.6份、热稳定剂2
‑
4份以及抗水解剂0.1
‑
1份。
64.作为一种实施方案,所述聚乙醇酸可以是乙醇酸均聚物,也可以是乙醇酸共聚物(例如,聚乙醇酸
‑
乳酸共聚物等)。
65.作为一种实施方案,所述柔性可降解树脂选自聚丁二酸乙二醇酯、聚丁二酸丁二醇酯、聚己二酸
‑
对苯二甲酸
‑
丁二醇酯、聚丁二酸
‑
对苯二甲酸
‑
丁二醇酯、聚丁二酸
‑
己二酸
‑
丁二醇酯、聚甲基乙撑碳酸酯、或聚己二酸乙二醇酯中的至少一种。
66.作为一种实施方案,所述相容剂选自eba
‑
g
‑
mah或eva
‑
g
‑
mah中的至少一种。所述eba
‑
g
‑
mah中ba含量为16
‑
18wt%,且熔融指数≤10g/10min(190℃,2.16kg),所述eva
‑
g
‑
mah中va含量为20
‑
28wt%,且熔融指数≤20g/10min(190℃,2.16kg)。
67.作为一种实施方案,所述增塑剂选自市售的甘油、环氧大豆油、环氧糠油酸丁酯或乙酰柠檬酸三丁酯中的至少一种。
68.作为一种实施方案,所述扩链剂为环氧类扩链剂,例如,可选自basf生产的adr 4370s。
69.作为一种实施方案,所述功能填料为95%过800目筛的改性沸石粉。
70.作为一种实施方案,所述抗氧剂选自β
‑
(3,5二叔丁基
‑4‑
羟基苯基)丙酸十八醇酯、1,1,3三(2
‑
甲基
‑
4羟基
‑
5叔丁基苯基)丁烷、2,2
’‑
甲撑双(4
‑
乙基
‑
6叔丁基苯酚)、1,3,
5
‑
三(3,5叔丁基
‑4‑
羟基苄基)三甲基苯、2,2
’‑
甲撑双(4
‑
甲基
‑6‑
叔丁基苯酚)或4,4
’‑
二叔辛基二苯胺中的至少一种。
71.作为一种实施方案,所述热稳定剂为有机锡类热稳定剂,优选为马来酸二丁基锡、二月桂酸二丁基锡、月桂酸马来酸二丁基锡、马来酸二正辛基锡、二月桂酸二正辛基锡或双(硫代甘醇酸异辛酯)二正辛基锡中的至少一种。
72.作为一种实施方案,所述抗水解剂为双(2,6
‑
二异丙基苯基)碳二亚胺。
73.可降解暂堵球的制备方法如下:
74.步骤1)熔融聚乙醇酸的制备:
75.步骤1
‑
1)将纯化的乙交酯粉末导入至熔融混合釜中,于常压下升温至110
‑
120℃,边搅拌边加入适量的反应助剂,使熔融的乙交酯与反应助剂混合均匀,获得呈流体态的预混料;
76.步骤1
‑
2)将呈流体态的预混料输送至静态混合器中进行预聚合,获得具有一定分子量的乙醇酸预聚物(例如,重均分子量为约5
‑
15万),随后将所述乙醇酸预聚物与交联剂一起输送至增粘设备中进行终聚合,以获得熔融聚乙醇酸;
77.步骤2)成型粒料的制备:按重量份将柔性可降解树脂、相容剂、增塑剂、扩链剂、功能填料、抗氧剂、热稳定剂和抗水解剂加入至混合挤出设备中,先进行塑化,获得塑化预混料,然后将增粘设备制得的熔融聚乙醇酸直接导入至混合挤出设备的共混段,使其与塑化预混料进行共混,再由混合挤出设备的挤出段挤出、造粒,即制得成型粒料;
78.步骤3)球体成型:将成型粒料置于注塑机中,加热至熔融状态,注射到具有设定直径的球形模具的内腔中,将球形模具自然冷却以消除残余应力,即制得可降解暂堵球。
79.在此需要说明的是,本发明中“增粘设备”起脱挥作用,可促使乙醇酸预聚物的进一步聚合,及时将生成的小分子脱除体系,以使聚合物的分子量得到进一步提升,对应地聚合物粘度也会进一步增大。本发明中使用的增粘设备可以为,例如,仅设有脱挥段的双螺杆挤出机。本发明中使用的混合挤出设备可以为,例如,设有塑化段、共混段及挤出段的双螺杆挤出机。
80.在上述制备方法中:
81.优选地,所述纯化的乙交酯粉末的d
90
≤200μm,纯度≥98.5%,酸度≤20mmol/kg。
82.优选地,所述反应助剂包含催化剂、引发剂及脱水剂。
83.在反应助剂用量方面,所述催化剂的用量为乙交酯质量的约0.001
‑
1wt%,所述引发剂的用量为乙交酯质量的约0.1
‑
1wt%,所述脱水剂的用量为乙交酯质量的约0.2
‑
1.6wt%。
84.优选地,所述催化剂可选自锡类化合物、锑类化合物或锌类化合物中的至少一种,例如但不限于辛酸亚锡、氯化亚锡、乳酸锡、三氧化二锑、二乙基锌或二水合乙酸锌。
85.所述引发剂可选自具有伯醇或仲醇等羟基结构的烷烃类物质(例如,正丙醇、异丙醇、正丁醇、异丁醇等)或具有羟基活性基团的芳香类物质(例如,苯甲醇、苯乙醇等)中的一种或两种。
86.优选地,所述脱水剂可选自碳化二亚胺、聚碳化二亚胺或基于碳化二亚胺的化合物(例如但不限于n,n
’‑
二异丙基碳二亚胺、二环己基碳二亚胺等)。
87.为防止出现熔融乙交酯局部反应助剂浓度过高,可采用现有的注射方式将反应助
剂逐滴加入至熔融混合釜中。
88.所述静态混合器中预聚合的温度控制为150
‑
230℃。
89.优选地,所述静态混合器可选自市售的sk型静态混合器。
90.优选地,所述静态混合器采用多段阶梯升温方式,例如,静态混合器共设有四段,第一段温度设定为150
‑
170℃,第二段温度设定为190
‑
210℃,第三段温度设定为210
‑
220℃,第四段温度设定为220
‑
230℃。
91.在实际制备过程中,物料在静态混合器中的总时长一般不超过150min,物料通过静态混合器的各段所需时间可以为:例如但不限于,通过第一段为约3
‑
8min,通过第二段为约10
‑
15min,通过第三段为约15
‑
20min,通过第四段为约50
‑
60min。
92.所述交联剂选自官能度≥3的多羟基烷烃类化合物(例如,双季戊四醇、丙三醇、三羟甲基乙烷、三羟甲基丙烷等)或含有不饱和烃的硅烷类化合物(例如,乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、乙烯基三(b
‑
甲氧基乙氧基)硅烷、γ
‑
(甲基丙烯酰氧)丙基三甲氧基硅烷等)中的至少一种。
93.在交联剂用量方面,所述交联剂的用量为基于乙交酯的质量计算所得聚乙醇酸理论质量的约1
‑
30wt%。
94.所述增粘设备仅设有脱挥段,该脱挥段的绝压≤200pa,且温度设定为220
‑
250℃,所述脱挥段的螺杆长径比设定为50
‑
60。
95.在实际生产过程中,所述交联剂可由脱挥段的起始处加入。
96.所述混合挤出设备中沿物料的进给方向依次设有塑化段、共混段及挤出段。
97.优选地,所述增粘设备的出料口与混合挤出设备中共混段的起始处相连接。
98.优选地,所述混合挤出设备中塑化段的温度控制为130
‑
160℃,共混段的温度控制为200
‑
220℃,挤出段的温度控制为220
‑
230℃。
99.优选地,所述混合挤出设备的螺杆长径比为40
‑
50。
100.上述增粘设备与混合挤出设备的连接关系如图1所示。
101.注塑机中,喂料温度控制为150
‑
160℃,计量段温度控制为190
‑
210℃,喷射口温度控制为230
‑
250℃,注塑压力控制为40
‑
90mpa,球形模具温度控制为120
‑
160℃。
102.以下为具体示例
103.实施例1
104.配方组分详见下表。
105.所采用的改性沸石由以下方法制备而成:
106.步骤i):将沸石粉加入至浓度为10wt%的naoh溶液中,于50℃的水浴中搅拌处理60min,后经固液分离,保留固体,并用去离子水洗涤至中性,再置于80℃的烘箱中干燥8h,即制得预处理沸石粉;
107.步骤ii):将预处理沸石粉加入至含有γ
‑
氨丙基三甲氧基硅烷和3
‑
(2,3
‑
环氧丙氧)丙基甲基二甲氧基硅烷的乙醇溶液(由无水乙醇和去离子水按质量比为1:5混合而成)中,于80℃的水浴中搅拌处理2h,制得悬浮液,再将悬浮液转移至超声波分散机,于35℃下采用400w超声处理2h,后经离心分离,保留固体,采用无水乙醇洗涤数次,于80℃的烘箱中干燥24h,研磨至95%过800目筛,即制得改性沸石粉。
108.上述步骤ii)中预处理沸石粉和乙醇溶液的用量关系为:每100g乙醇溶液中加入
10g预处理沸石粉,并且γ
‑
氨丙基三甲氧基硅烷的用量为预处理沸石粉质量的1.2%,而3
‑
(2,3
‑
环氧丙氧)丙基甲基二甲氧基硅烷的用量为预处理沸石粉质量的0.6%。
109.实施例2
110.配方组分详见下表。
111.所采用的改性沸石由以下方法制备而成:
112.步骤i):将沸石粉加入至浓度为25wt%的naoh溶液中,于60℃的水浴中搅拌处理20min,后经离心分离,保留固体,并用去离子水洗涤至中性,再置于80℃的烘箱中干燥8h,即制得预处理沸石粉;
113.步骤ii):将预处理沸石粉加入至含有γ
‑
氨丙基三甲氧基硅烷和3
‑
(2,3
‑
环氧丙氧)丙基甲基二甲氧基硅烷的乙醇溶液(由无水乙醇和去离子水按质量比为1:1混合而成)中,于90℃的水浴中搅拌处理1h,制得悬浮液,再将悬浮液转移至超声波分散机,于60℃下采用600w超声处理1h,后经离心分离,保留固体,采用无水乙醇洗涤数次,于100℃的烘箱中干燥10h,研磨至95%过800目筛,即制得改性沸石粉。
114.上述步骤ii)中预处理沸石粉和乙醇溶液的用量关系为:每100g乙醇溶液中加入25g预处理沸石粉,并且γ
‑
氨丙基三甲氧基硅烷的用量为预处理沸石粉质量的1.8%,而3
‑
(2,3
‑
环氧丙氧)丙基甲基二甲氧基硅烷的用量为预处理沸石粉质量的0.8%。
115.实施例3、4
116.配方组分详见下表。
117.所采用的改性沸石由以下方法制备而成:
118.步骤i):将沸石粉加入至浓度为20wt%的naoh溶液中,于56℃的水浴中搅拌处理35min,后经离心分离,保留固体,并用去离子水洗涤至中性,再置于100℃的烘箱中干燥6h,即制得预处理沸石粉;
119.步骤ii):将预处理沸石粉加入至含有γ
‑
氨丙基三甲氧基硅烷和3
‑
(2,3
‑
环氧丙氧)丙基甲基二甲氧基硅烷的乙醇溶液(由无水乙醇和去离子水按质量比为1:3混合而成)中,于86℃的水浴中搅拌处理2h,制得悬浮液,再将悬浮液转移至超声波分散机,于50℃下采用500w超声处理2h,后经离心分离,保留固体,采用无水乙醇洗涤数次,于100℃的烘箱中干燥12h,研磨至95%过800目筛,即制得改性沸石粉。
120.上述步骤ii)中预处理沸石粉和乙醇溶液的用量关系为:每100g乙醇溶液中加入20g预处理沸石粉,并且γ
‑
氨丙基三甲氧基硅烷的用量为预处理沸石粉质量的1.6%,而3
‑
(2,3
‑
环氧丙氧)丙基甲基二甲氧基硅烷的用量为预处理沸石粉质量的0.4%。
121.实施例1
‑
4中各组分及其用量关系如下表1所示。
122.表1配方组分及其重量份
123.项目聚乙醇酸柔性可降解树脂相容剂增塑剂扩链剂功能填料抗氧剂热稳定剂抗水解剂实施例17030420.5150.220.1实施例27525531200.820.4实施例38218631.3241.230.6实施例49010852301.641
124.实施例1
‑
4中各组分及其种类如下表2
‑
1和表2
‑
2所示:
125.表2
‑
1配方组分及其种类
[0126][0127]
注:表2
‑
1中柔性可降解树脂的重均分子量为约6万。
[0128]
表2
‑
2配方组分及其种类
[0129][0130][0131]
依据上述实施例1
‑
4,采用以下方法来制备暂堵球:
[0132]
步骤1)熔融聚乙醇酸的制备:
[0133]
步骤1
‑
1)将经纯化的乙交酯粉末(粒径d
90
≤200μm,纯度≥98.5%,酸度≤20mmol/kg)导入至熔融混合釜中,于常压下升温至110
‑
120℃(例如,实施例1为约110℃,实施例2为约112℃,实施例3为约116℃,实施例4为约120℃),边搅拌边加入适量的反应助剂,使熔融的乙交酯与反应助剂混合均匀,获得呈流体态的预混料;
[0134]
步骤1
‑
2)将呈流体态的预混料输送至静态混合器中进行预聚合,获得具有一定分子量的乙醇酸预聚物,随后将所述乙醇酸预聚物与交联剂一起输送至增粘设备中进行终聚合,以获得熔融聚乙醇酸;
[0135]
步骤2)成型粒料的制备:按重量份将柔性可降解树脂、相容剂、增塑剂、扩链剂、功能填料、抗氧剂、热稳定剂和抗水解剂加入至混合挤出设备中,先进行塑化,获得塑化预混料,然后将增粘设备制得的熔融聚乙醇酸直接导入至混合挤出设备的共混段,使其与塑化预混料进行共混,再由混合挤出设备的挤出段挤出、造粒,即制得成型粒料;
[0136]
步骤3)球体成型:将成型粒料置于注塑机中,加热至熔融状态,注射到具有设定直径(例如d=19mm)的球形模具的内腔中,待完成注射后,将球形模具自然冷却至80
‑
100℃(例如90℃),保温1
‑
2小时(例如2小时),以消除残余应力,随后再自然冷却至室温,即制得可降解暂堵球。
[0137]
上述步骤1
‑
1)中,反应助剂的添加量如下表3
‑
1所示:
[0138]
表3
‑
1反应助剂的添加量
[0139][0140]
上述步骤1
‑
2)中,反应助剂的添加量如下表3
‑
2所示:
[0141]
表3
‑
2交联剂的添加量
[0142][0143]
上述步骤1
‑
1)和1
‑
2)中所使用的反应助剂和交联剂的种类如下表3
‑
3所示:
[0144]
表3
‑
3反应助剂和交联剂的种类
[0145][0146][0147]
上述步骤1
‑
2)中静态混合器的工艺条件如下表4
‑
1和表4
‑
2所示:
[0148]
表4
‑
1静态混合器各段温度参数
[0149]
项目第一段第二段第三段第四段实施例1约150℃约190℃约210℃约220℃实施例2约156℃约200℃约215℃约222℃实施例3约164℃约204℃约218℃约226℃实施例4约170℃约210℃约220℃约230℃
[0150]
表4
‑
2物料通过静态混合器各段所需的时间
[0151]
项目第一段第二段第三段第四段实施例1约5min约15min约20min约50min实施例2约5min约10min约15min约60min实施例3约8min约10min约16min约56min实施例4约3min约12min约20min约55min
[0152]
上述步骤1
‑
2)中i类双螺杆挤出机中脱挥段的参数设定如下表5所示:
[0153]
表5 i类双螺杆挤出机中脱挥段的参数设定
[0154]
[0155][0156]
注:表5中物料通过脱挥段的时间为约15min.
[0157]
◆
分子量及分子量分布的测试
[0158]
取0.2g pga样品,溶于三氟乙酸钠含量为5mmol/l的六氟异丙醇溶液100ml,经过0.4μm孔径的聚四氟乙烯滤膜过滤,取20μl加入到岛津(日本)制“lc
‑
20ad gpc”进样器中,测试条件:柱温40℃;洗脱液:溶解有5mmol/l三氟乙酸钠的六氟异丙醇;流速1ml/min;检测器:ri检测器;校正:使用分子量在7000至200000不等的五种不同标准聚甲基烯酸甲酯进行分子量校正。
[0159]
上述步骤1
‑
2)中所得乙醇酸预聚物、熔融聚乙醇酸的分子量及其分布测试结果如下表6所示:
[0160]
表6分子量及分子量分布测试结果
[0161][0162]
上述步骤2)中混合挤出设备中各段的参数设定如下表7所示:
[0163]
表7混合挤出设备中各段的参数设定
[0164][0165]
上述步骤3)中注塑机和球形模具的参数设定如下表8所示:
[0166]
表8注塑机中各段的参数设定
[0167][0168]
下面提供对比例:
[0169]
对比例1:
[0170]
本对比例中采用普通市售的沸石粉(95%过800目筛)来替代功能填料,其余同实施例3。
[0171]
对比例2:
[0172]
本对比例中所使用的聚乙醇酸是由乙交酯粉末(d
90
≤200μm,纯度≥98.5%,酸度≤20mmol/kg)经由常规反应型双螺杆挤出机挤出、造粒而制得,所使用的反应助剂与上述实施例3相同,具体工艺条件及参数如下表9
‑
1和表9
‑
2所示。
[0173]
表9
‑
1常规反应型双螺杆挤出机的参数设定
[0174][0175]
注:对比例2中乙交酯粉末与反应助剂混合均匀后,由反应型双螺杆挤出机的混合段的第一段起始处加入。
[0176]
表9
‑
2常规反应型双螺杆挤出机的参数设定
[0177][0178]
本对比例中基于常规反应型双螺杆挤出机制得的聚乙醇酸的数均分子量(mn)为约90128,重均分子量(mw)为约158626,分子量分布指数为约1.76。
[0179]
按实施例3配方中各组分的用量关系,将制得的聚乙醇酸和柔性可降解树脂由双螺杆挤出机的主喂料口加入,随后将相容剂、增塑剂、扩链剂、功能填料、抗氧剂、热稳定剂和抗水解剂由双螺杆挤出机的侧喂料口加入,控制双螺杆挤出机的塑化段温度为约206℃,共混段温度为约220℃,挤出段温度为230℃,后经挤出、造粒,即制得成型粒料。
[0180]
将制得的成型粒料置于注塑机中,加热至熔融状态,注射到具有设定直径的球形
模具的内腔中,将球形模具自然冷却以消除残余应力,即制得可降解暂堵球。
[0181]
本对比例中,注塑机和球形模具的相关参数设置与实施例3相同。
[0182]
对比例3:
[0183]
本对比例所采用的聚乙醇酸为对比例2中由常规反应型双螺杆挤出机经挤出、造粒而制成,其作为成型粒料,不含有其它加工助剂。
[0184]
将成型粒料置于注塑机中,加热至熔融状态,注射到具有设定直径的球形模具的内腔中,将球形模具自然冷却以消除残余应力,即制得可降解暂堵球。
[0185]
本对比例中,注塑机和球形模具的相关参数设置与实施例3相同。
[0186]
性能测试:
[0187]
将上述实施例1
‑
4和对比例1
‑
3制得的暂堵球(直径为19mm)进行以下步骤的水中降解测试:
[0188]
步骤(1):取4个暂堵球,置于恒温干燥箱中,于105℃下干燥2小时,称量,记录初始质量为m0;
[0189]
步骤(2):将干燥后的4个暂堵球分别置于一端开口的硬质玻璃器皿内,再分别加入适量的清水以完全浸泡暂堵球,将4个硬质玻璃器皿分别标记为s1、s2、s3和s4,随后置于恒温恒湿试验箱中,将温度设定为90℃;
[0190]
步骤(3):待经过8小时后,取出s1中的暂堵球,将其表面的碎片剥离后,用蒸馏水清洗干净,并放入恒温干燥箱,于105℃下烘干2小时后称重,记录剩余质量为m1;
[0191]
步骤(4):待经过1天后,取出s2中的暂堵球,将其表面的碎片剥离后,用蒸馏水清洗干净,并放入恒温干燥箱,于105℃下烘干2小时后称重,记录剩余质量为m2;
[0192]
步骤(5):待经过3天后,取出s3中的暂堵球,将其表面的碎片剥离后,用蒸馏水清洗干净,并放入恒温干燥箱,于105℃下烘干2小时后称重,记录剩余质量为m3;
[0193]
步骤(6):待经过5天后,取出s4中的暂堵球,将其表面的碎片剥离后,用蒸馏水清洗干净,并放入恒温干燥箱,于105℃下烘干2小时后称重,记录剩余质量为m4。
[0194]
依据以下计算公式来计算暂堵球的降解率r
d
:
[0195]
r
d
(s1)=(m0‑
m1)/m0×
100%;
[0196]
r
d
(s2)=(m0‑
m2)/m0×
100%;
[0197]
r
d
(s3)=(m0‑
m3)/m0×
100%;
[0198]
r
d
(s4)=(m0‑
m4)/m0×
100%。
[0199]
在上述测试步骤中,在进行质量测量时,针对暂堵球已基本消失的情况,可采用以下方式来进行质量测量:取出硬质玻璃器皿,抽取上层清液以分离剩余固相,并将分离的剩余固相用蒸馏水清洗干净,并放入恒温干燥箱,在105℃条件下烘干2小时后称重,记录剩余固相质量。
[0200]
在实际测量过程中,为确保测量的准确性,可重复上述方法若干次,记录相应的测试结果,并可对测试结果取平均值。
[0201]
实施例1
‑
4和对比例1
‑
3制得的暂堵球的水中降解测试结果如下表10所示:
[0202]
表10暂堵球的水中降解测试结果
[0203]
项目经过8小时(降解率)经过1天(降解率)经过3天(降解率)经过5天(降解率)实施例1约5.3%约26.3%约50.6%约88.2%
实施例2约4.2%约23.5%约48.3%约84.7%实施例3约3.1%约16.4%约41.8%约75.6%实施例4约2.9%约18.7%约44.7%约78.4%对比例1约4.7%约25.6%约66.8%约81.2%对比例2约6.3%约27.8%约62.6%约79.3%对比例3约9.5%约38.4%约82.9%约94.5%
[0204]
将上述实施例1
‑
4和对比例1
‑
3制得的暂堵球(直径为19mm)进行以下步骤的封堵承压测试:
[0205]
①
将待测试的暂堵球坐封于直径为约9mm的球座上,密封完全,设置测试温度为90℃,恒定后从20mpa起逐级开始打压,暂堵球无破碎,继续加压直至暂堵球被击穿压力降为零,在此之前的最高压力即为暂堵球在90℃下的承压强度。
[0206]
以上承压测试过程中,每个产品需平行测试5组,取算数平均值,测试结果见表11
‑
1所示:
[0207]
表11
‑
1封堵承压测试结果
[0208]
项目实施例1实施例2实施例3实施例4对比例1对比例2对比例3承压强度/(mpa)约76约79约87约85约72约70约54
[0209]
②
将待测试的暂堵球坐封于直径为9mm的球座上,密封完全,设置测试温度为90℃,恒定后施加70mpa的压力,保持压力恒定,直至暂堵球无法维持稳定的承压,压力骤降幅度超过5%,即停止测试,记录暂堵球的稳压时间。
[0210]
以上承压测试过程中,每个产品需平行测试5组,取算数平均值,测试结果见表11
‑
2所示:
[0211]
表11
‑
2封堵承压测试结果
[0212]
项目实施例1实施例2实施例3实施例4对比例1对比例2对比例370mpa下的稳压时间(h)约17约21约30约27约12约9*
[0213]
注:表11
‑
2中稳压时间的测量是以小时来计,采用四舍五入来记录时间值,另,符号“*”表示无法正常测量。
[0214]
将实施例3和对比例1
‑
3制得的暂堵球(直径为约19mm)于25℃、50mpa的压力条件下,进行弹性变形性能测试,测试结果如下表12所示:
[0215]
表12弹性变形性能测试结果
[0216]
项目变形前横向直径变形时横向直径变形恢复后横向直径变形率恢复率实施例319.04mm20.78mm19.31mm9.14%84.48%对比例118.95mm20.13mm19.34mm6.23%66.95%对比例219.12mm20.94mm19.59mm9.52%74.18%对比例319.07mm19.86mm19.56mm4.14%37.97%
[0217]
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1