一种紫色活性染料的制备工艺的制作方法
1.本发明属于活性染料合成领域,具体涉及一种紫色活性染料的制备工艺。
背景技术:
2.活性染料是染料行业的重要分支,该类染料主要由染料母体、连接基以及活性基团构成,其活性基团能够与棉、毛、麻等的纤维素纤维发生亲核取代反应,形成稳定的共价键,故又称反应性染料。自上世纪50年代,活性染料出现开始,人们对于活性染料的开发与探索从未停止,随着研究深入,活性染料的优点不断被挖掘,包括色泽鲜艳、色谱广泛、溶解度高、染色工艺简单、适用范围广等等,逐渐适应市场上大部分纤维和布料的要求。
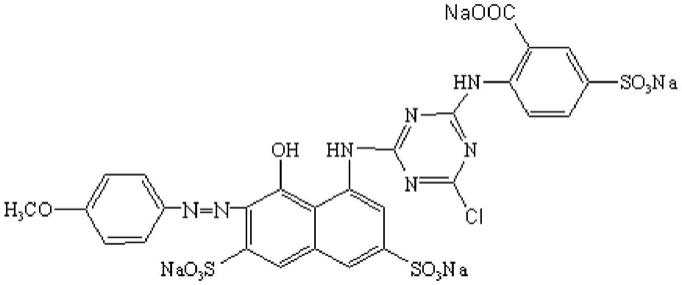
3.c.i.reactive violet 46,中文别名活性紫46、活性紫rrn,分子式 c
27h16
cln7na4o
13
s3,分子量870.07,是紫色活性印花染料的代表之一,可直接应用于棉、锦纶、丝绸、羊毛等织物的印花,也可进一步深加工成液体染料及喷墨印花染料,提高产品的附加值。
4.结构式如下:
[0005][0006]
传统活性紫rrn的合成方法为对氨基苯甲醚重氮盐与h酸溶液碱性偶合得到偶合液,偶合液加入氯化钠、氯化钾盐析精制,得到色基,色基再与三聚氯氰进行一次缩合,然后加入2-氨基-5-磺酸基苯甲酸进行二次缩合,最后加入氯化钾盐析,压滤得到滤饼,溶解滤饼得到原浆。该传统工艺优势在于,色光稳定,且能够在一定范围内控制原浆含固量,减少喷干成本。但劣势也很明显,首先,需要经过两步盐析工序,产生大量高盐分、高cod有色废水,同时盐析损耗大,产品收量不高,原料成本高居不下;其次,为保证反应稳定进行,使用磷酸钠的一种或两种调配成缓冲盐参与缩合反应,合成的产品在印染过程会产生含磷废水,面料上也会有磷元素残留;再次,染料溶解度不高,一般不超过100g/l,深加工成液体染料过程中需要加入大量助溶剂,增加成本,且储存稳定性差,长时间放置有稠状物析出、固色率下降等风险;最后,生产周期长,每批次的产品生产需要花费的时间都在65h以上,不论是色基制备还是盐析精制都需要耗费大量的人力物力,占用生产资源,限制公司产能。
[0007]
国内外市场分析:对低端产品基本选用乙烯砜结构的活性紫,如c.i.reactive violet 5,中文别名活性紫5、活性紫kn-4r,乙烯砜结构的活性紫印花色浆糊料在制备、贮藏过程会水解,在印花过程中在织物上染料的固色率低,也极易造成色花;对高端产品乙烯
砜结构无法满足市场需求,传统工艺制造的紫rrn成本高,与国外同类产品竞争处于劣势,基本靠进口紫 rrn来维持该领域的需求。
技术实现要素:
[0008]
本发明的目的在于针对现有技术的不足,提供一种紫色活性染料的制备工艺,该制备工艺可以减少传统盐析精制法产生的大量高cod、高盐分的有色废水,同时合成的染料具有良好的溶解度,染色牢度好、固色率高、布面无磷残留且色泽鲜艳,热稳定性表现优异和加工成液体染料储存性能好。
[0009]
本发明所提供的技术方案为:
[0010]
一种紫色活性印花染料的制备工艺,包括如下步骤:
[0011]
1)将h酸单钠盐投加到水中打浆,调节ph值在6.5-7.2,搅拌溶解,得到h酸溶清液;
[0012]
2)三聚氯氰冰浴打浆分散后,滴加步骤1)得到的h酸溶清液,控制温度3-8℃,ph值在1.5-1.8,进行一次缩合反应,反应完全后得到一缩液;
[0013]
3)在混酸存在的条件下,对氨基苯甲醚与亚硝酸钠进行重氮化反应,反应完全后得到重氮盐溶液;
[0014]
4)将步骤3)得到的重氮液加入步骤2)得到的一缩液中进行偶合反应,控制温度2-6℃,调节ph值在6.8-7.0,反应完全后得到偶合液;
[0015]
5)2-氨基-5-磺酸基苯甲酸加水,调节ph值在7.0-7.5,搅拌溶解,得到中和液;
[0016]
6)向步骤4)得到的偶合液中加入步骤5)得到的中和液,升温至50-55℃,维持ph值在7.3-7.5进行二次缩合反应,反应完全后即得紫色染料原浆。
[0017]
所述步骤1)中h酸单钠盐溶解反应过程如下:
[0018][0019]
本发明步骤1)中h酸单钠盐极难溶于水,使用调碱的方式,先将h酸溶清,再参与到一缩反应中,可以提高物料接触效果,提高转化率,缩短反应时间。
[0020]
作为优选,所述步骤1)中,使用工业级30%液碱调节ph值,得到的h 酸溶清液控制质量分数在25-30%。
[0021]
作为优选,所述步骤1)中,溶解完成后,降温并保持温度2-6℃。
[0022]
所述步骤2)中一次缩合反应反应过程如下:
[0023]
[0024]
本发明步骤2)中,通过检测ph值把控打浆进度,ph值处于标定范围内时表示打浆完成;三聚氯氰与h酸缩合反应实际为本产品第一步合成反应,空间位阻较小,反应较易进行,无需使用缓冲盐,使用小苏打调节即可稳定反应,另外一次缩合反应是一个先慢后快再慢的过程,所以在实际滴加h酸溶清液的过程中,需要控制滴加节奏,保证反应处于最佳的状态,同时控制反应温度,防止局部过热导致三聚氯氰水解;最后,控制好反应时间,并以 hplc检测来确定反应终点。
[0025]
作为优选,所述步骤2)中,三聚氯氰打浆后,ph值在2.0-2.5时,打浆效果最佳,开始滴加h酸溶清液。
[0026]
作为优选,所述步骤2)中,h酸溶清液总滴加时间为75-90min,前 25-30min控制滴加量为h酸溶清液总体积的1/4,中间25-30min控制滴加量为h酸溶清液总体积的1/2,最后25-30min控制滴加量为h酸溶清液总体积的1/4。
[0027]
作为优选,所述步骤2)中,h酸溶清液的滴加和反应过程,使用小苏打维持ph值,所述小苏打与h酸摩尔比为(0.9-1.2):1。
[0028]
作为优选,所述步骤2)中,反应时间控制在3-4h,反应终点为hplc 检测一缩物纯度》97%。
[0029]
所述步骤3)中重氮反应反应过程如下:
[0030][0031][0032]
本发明步骤3)中,重氮反应需要在强酸性条件下进行,采用醋酸代替一部分盐酸组成混酸参与到重氮反应中,醋酸对对氨基苯甲醚有较好的溶解作用,使得对氨基苯甲醚更亲于水相,更有利于与亚硝酸钠进行重氮反应,提高重氮反应的收率;也为后续反应增加了醋酸-醋酸钠缓冲体系。该缓冲体系可以替代磷酸盐在二次缩合反应发挥维稳作用,也能够在染料的储存、印染过程中抑制水解,提高稳定性。
[0033]
作为优选,所述步骤3)中,混酸为盐酸和醋酸混合液。
[0034]
作为优选,所述步骤3)中盐酸、醋酸、亚硝酸钠与对氨基苯甲醚的摩尔比为(0.5-1.0):(0.5-1.0):(1.0-1.05):1。
[0035]
作为优选,所述步骤3)中重氮化反应温度控制为0-5℃,反应时间控制在4-5h。
[0036]
所述步骤4)中偶合反应的反应过程如下:
[0037]
[0038]
本发明步骤4)中,偶合反应主要发生在h酸的氨基邻位和羟基邻位,本产品需要h酸羟基邻位的偶合产物。为此,在偶合前向一缩液中加入纯碱,降低h酸氨基邻位的活性,同时在滴加重氮盐过程中维持好ph值,重氮盐能够快速在h酸的羟基邻位偶合,减少了偶合时间,副产减少,偶合液纯度提高,染料相对固色率提升。若使用小苏打来调节ph值,则偶合收率会降低 10-15%,原因在于小苏打的碱性太弱,偶合速率太慢会导致部分重氮盐水解;若用液碱或稀液碱来调节ph值,则偶合收率会降低5-8%,相对固色率降低 10-12%,收率低的原因在于使用强碱后偶合对与调节ph值过程中无缓冲体系,使得强碱直接与重氮盐接触,而失去偶合能力,造成收率偏低;相对固色率低的原因在于体系缓冲能力差,一缩物碰到强碱会引起结构中第二个氯水解成羟基,后续无法参与二次缩合,造成相对固色率下降。
[0039]
作为优选,所述步骤4)中重氮液加入一缩液前,先向一缩液中加入工艺量纯碱。
[0040]
进一步优选,所述步骤4)中重氮液加入一缩液前,先向一缩液中加入纯碱,纯碱质量为三聚氯氰的10-15%。
[0041]
作为优选,所述步骤4)中重氮液加入一缩液时间控制在20-30min,加入过程中控制ph值大于6.5,温度小于6℃。
[0042]
作为优选,所述步骤4)中重氮液加完后,调配20%纯碱液维持ph值,维持反应时间为2-3h。
[0043]
所述步骤5)中2-氨基-5-磺酸基苯甲酸中和反应的反应过程如下:
[0044][0045]
作为优选,所述步骤5)中使用工业级30%液碱来调ph,得到的2-氨基-5-磺酸基苯甲酸中和液质量分数在20-30%。
[0046]
所述步骤6)中二次缩合反应反应过程如下:
[0047][0048]
作为优选,所述步骤6)中中和液加完后,调配20%纯碱液维持ph值,维持反应时间为4-5h。
[0049]
作为优选,所述步骤1)到步骤6)中,三聚氯氰、h酸单钠盐、对氨基苯甲醚与2-氨基-5-磺酸基苯甲酸的摩尔比为1:(0.98-1.03):(0.97-1.02): (0.96-1.01)。
[0050]
同现有技术相比,本发明的有益效果在于:
[0051]
(1)优化反应流程,省去色基的盐析和二缩液的盐析,收率提高,同时减少高cod有色废水排放,降低生化降解压力,绿色环保;
[0052]
(2)生产周期短,能耗低,能源使用效率高;
[0053]
(3)固色率高、溶解度好,可以加工成高浓度液体染料,产品回报率高;
[0054]
(4)性能稳定,便于储存,产品热稳定性检测表现优异;
[0055]
(5)染料无磷化,应用后不会导致水体富营养化。
具体实施方式
[0056]
实施例1:
[0057]
5方反应锅中加底水1000kg,投入h酸单钠盐430kg,搅拌30min,滴加 30%工业液碱140kg,调节ph值在6.5-7.2,h酸溶解,得到溶清液,降温至 2-6℃,保温备用。
[0058]
15方反应锅中加底水600kg、碎冰1000kg,投入三聚氯氰200kg,搅拌打浆,ph值在2.0-2.5时,测量备用的h酸溶清液体积,然后开始滴加h酸溶清液,控制滴加节奏,前25-30min控制滴加量为h酸溶清液总体积的1/4, 中间25-30min控制滴加量为h酸溶清液总体积的1/2,最后25-30min控制滴加量为h酸溶清液总体积的1/4,滴加过程用小苏打保持ph值在1.5-1.8,滴完后,继续用小苏打维持ph值在1.5-1.8,保温3-8℃,反应3-4h,hplc检测一缩物纯度》97%时,反应终点到,得到一缩液。
[0059]
5方重氮锅中加底水600kg,升温至40℃,投入对氨基苯甲醚125kg,搅拌20min,加入30%工业盐酸100kg、99.5%工业醋酸40kg,继续搅拌10min,加碎冰降温至0-5℃,滴加
30%亚硝酸钠溶液250kg,滴完保温0-5℃并继续反应4-5h,用氨基磺酸消除过量的亚硝酸钠,得到对氨基苯甲醚重氮盐备用。
[0060]
一缩液中加入纯碱25kg,将5方重氮锅中对氨基苯甲醚重氮盐转移到一缩液内,转移时间控制在20-30min,ph值大于6.5,温度小于6℃,转移完成后继续用20%纯碱液维持ph值在6.8-7.0,保温2-6℃,反应2-3h,反应完全后得到偶合液。
[0061]
5方反应锅中加底水500kg,投入2-氨基-5-磺酸基苯甲酸230kg,搅拌 30min,滴加30%工业液碱285kg,调节ph值在7.0-7.5,搅拌溶解,得到中和液。
[0062]
将2-氨基-5-磺酸基苯甲酸中和液快速转移到偶合液中,升温至50-55℃,用20%纯碱液维持ph值在7.3-7.5,继续保温50-55℃,反应4-5h,反应完全后即得紫色染料原浆。
[0063]
将原浆标准化后,喷雾干燥,得到活性紫rrn染料干粉。
[0064]
实施例2:
[0065]
5方反应锅中加底水1000kg,投入h酸单钠盐440kg,搅拌30min,滴加 30%工业液碱143kg,调节ph值在6.5-7.2,h酸溶解,得到溶清液,降温至 2-6℃,保温备用。
[0066]
15方反应锅中加底水600kg、碎冰1000kg,投入三聚氯氰200kg,搅拌打浆,ph值在2.0-2.5时,测量备用的h酸溶清液体积,然后开始滴加h酸溶清液,控制滴加节奏,前25-30min控制滴加量为h酸溶清液总体积的1/4, 中间25-30min控制滴加量为h酸溶清液总体积的1/2,最后25-30min控制滴加量为h酸溶清液总体积的1/4,滴加过程用小苏打保持ph值在1.5-1.8,滴完后,继续用小苏打维持ph值在1.5-1.8,保温3-8℃,反应3-4h,hplc检测一缩物纯度》97%时,反应终点到,得到一缩液。
[0067]
5方重氮锅中加底水600kg,升温至40℃,投入对氨基苯甲醚128kg,搅拌20min,加入30%工业盐酸110kg、99.5%工业醋酸35kg,继续搅拌10min,加碎冰降温至0-5℃,滴加30%亚硝酸钠溶液256kg,滴完保温0-5℃并继续反应4-5h,用氨基磺酸消除过量的亚硝酸钠,得到对氨基苯甲醚重氮盐备用。
[0068]
一缩液中加入纯碱25kg,将5方重氮锅中对氨基苯甲醚重氮盐转移到一缩液内,转移时间控制在20-30min,ph值大于6.5,温度小于6℃,转移完成后继续用20%纯碱液维持ph值在6.8-7.0,保温2-6℃,反应2-3h,反应完全后得到偶合液。
[0069]
5方反应锅中加底水500kg,投入2-氨基-5-磺酸基苯甲酸232kg,搅拌 30min,滴加30%工业液碱290kg,调节ph值在7.0-7.5,搅拌溶解,得到中和液。
[0070]
将2-氨基-5-磺酸基苯甲酸中和液快速转移到偶合液中,升温至50-55℃,用20%纯碱液维持ph值在7.3-7.5,继续保温50-55℃,反应4-5h,反应完全后即得紫色染料原浆。
[0071]
将原浆标准化后,喷雾干燥,得到活性紫rrn染料干粉。
[0072]
实施例3:
[0073]
5方反应锅中加底水1000kg,投入h酸单钠盐430kg,搅拌30min,滴加 30%工业液碱140kg,调节ph值在6.5-7.2,h酸溶解,得到溶清液,降温至 2-6℃,保温备用。
[0074]
15方反应锅中加底水600kg、碎冰1000kg,投入三聚氯氰200kg,搅拌打浆,ph值在2.0-2.5时,测量备用的h酸溶清液体积,然后开始滴加h酸溶清液,控制滴加节奏,前25-30min控制滴加量为h酸溶清液总体积的1/4, 中间25-30min控制滴加量为h酸溶清液总体积的1/2,最后25-30min控制滴加量为h酸溶清液总体积的1/4,滴加过程用小苏打保持ph值在1.5-1.8,滴完后,继续用小苏打维持ph值在1.5-1.8,保温3-8℃,反应3-4h,hplc检测一
缩物纯度》97%时,反应终点到,得到一缩液。
[0075]
5方重氮锅中加底水600kg,升温至40℃,投入对氨基苯甲醚125kg,搅拌20min,加入30%工业盐酸100kg、99.5%工业醋酸40kg,继续搅拌10min,加碎冰降温至0-5℃,滴加30%亚硝酸钠溶液250kg,滴完保温0-5℃并继续反应4-5h,用氨基磺酸消除过量的亚硝酸钠,得到对氨基苯甲醚重氮盐备用。
[0076]
一缩液中加入纯碱28kg,将5方重氮锅中对氨基苯甲醚重氮盐转移到一缩液内,转移时间控制在20-30min,ph值大于6.5,温度小于6℃,转移完成后继续用20%纯碱液维持ph值在6.8-7.0,保温2-6℃,反应2-3h,反应完全后得到偶合液。
[0077]
5方反应锅中加底水500kg,投入2-氨基-5-磺酸基苯甲酸230kg,搅拌 30min,滴加30%工业液碱285kg,调节ph值在7.0-7.5,搅拌溶解,得到中和液。
[0078]
将2-氨基-5-磺酸基苯甲酸中和液快速转移到偶合液中,升温至50-55℃,用20%纯碱液维持ph值在7.3-7.5,继续保温50-55℃,反应4-5h,反应完全后即得紫色染料原浆。
[0079]
将原浆标准化后,喷雾干燥,得到活性紫rrn染料干粉。
[0080]
对比例1:
[0081]
5方重氮锅中加底水600kg,升温至40℃,投入对氨基苯甲醚125kg,搅拌20min,加入30%工业盐酸160kg,继续搅拌10min,加碎冰降温至0-5℃,滴加30%亚硝酸钠溶液245kg,滴完保温3-8℃并继续反应4-5h,用氨基磺酸消除过量的亚硝酸钠,得到对氨基苯甲醚重氮盐备用。
[0082]
5方反应锅中加底水900kg,投入h酸单钠盐420kg,搅拌30min,滴加 30%工业液碱138kg,调节ph值在6.5-7.2,h酸溶解,得到溶清液备用。
[0083]
将h酸溶清液转移到对氨基苯甲醚重氮盐中,转移完成后,开始使用小苏打调节ph值6-6.5,降温至5-10℃,保温反应6-7h。终点到后,升温至 45-50℃,测量体积,先投入10%体积比nacl,再投入10%体积比kcl,低速搅拌,3-4h后大量物料析出,定性滤纸上点斑点清晰且渗圈干净,压滤吹干,搜集滤饼,5方反应锅加底水800kg,投入滤饼
①
,搅拌溶解,得到色基,加入磷酸二氢钠10kg、磷酸氢二钠20kg,备用。
[0084]
15方反应锅中加底水600kg、碎冰1000kg,投入三聚氯氰200kg,搅拌打浆2h,加入色基,加完后用小苏打调节ph值在5.5-6,保温10-15℃,反应6-7h。一缩终点到后,直接加入2-氨基-5-磺酸基苯甲酸232kg、磷酸二氢钠 10kg、磷酸氢二钠20kg,开始升温,保温50-55℃,用15%工业液碱维持ph 值在7.5-8.0,反应4-5h。二缩终点到后,测量体积,加入18%体积比kcl,低速搅拌,3-4h后大量染料析出,定性滤纸上点斑点清晰且渗圈浅色,压滤吹干,搜集滤饼
②
。
[0085]
15方反应锅加底水6000kg,投入滤饼
②
,搅拌,并用少量工业盐酸回调 ph值在6.5-7.0,即得紫色染料原浆。
[0086]
将原浆标准化后,喷雾干燥,得到活性紫rrn染料干粉。
[0087]
对比例2:
[0088]
按染料生产工艺汇编(编制:上海市有机化学工业公司,1976年2月) 中第306页活性艳紫kn-4r合成工艺,制备乙烯砜结构活性紫染料,得到对比2样品
[0089]
将实施例1-3分别制备的紫色活性染料产品,与按传统工艺合成的对比例 1紫色活性染料产品和已公开的乙烯砜结构紫对比例2相比较,表1对比色光、相对强度、相对固色
率、相对收率与溶解度(以企标为标准);表2对标准化的样品在72℃环境下存放3天,相对固色率的变化,及配制成相同浓度的印花色浆料在35℃下对比第1、3、5、7天的相对固色率。
[0090]
对比例3:
[0091]
实施例1,重氮工序中不使用醋酸,醋酸等摩尔量的酸用盐酸代替,其它均不变。
[0092]
对比例4:
[0093]
实施例1,偶合工序中不使纯碱,用15-30%液碱替代纯碱,其它均不变。
[0094]
表1
[0095][0096][0097]
表2
[0098]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1