一种改性的g‑C3N4基可见光光催化剂、其制备方法和应用与流程
本发明涉及光催化材料领域,特别涉及对g-c3n4改性后的g-c3n4基光催化剂和制备方法。
背景技术:
自1989年加州大学的liu和cohen从理论上提出β-c3n4共价晶体以来,碳化氮化合物因此在光催化领域受到各国科学家的广泛关注,1989年a.y.liu和m.l.cohen根据β-si3n4的晶体结构,用c替换si,在局域态密度近似下采用第一性赝势能带法从理论上预言了β-c3n4(即氮化碳)这种硬度可以和金刚石相媲美而且是在自然界中尚未发现的新的共价化合物。1996年,teter和hemley通过计算认为c3n4可能具有5种结构,即α相,β相,立方相、准立方相以及类石墨相,其中石墨相c3n4是常温下c3n4中最稳定的相,同时材料无毒,并且有可见光响应,因此被广泛应用于光催化中。除此之外c3n4制备简单、价格便宜、不含金属使其成为一种新型光催化剂。
但g-c3n4(石墨相碳化氮)也存在着一些缺点,比如光生电子-空穴复合率高而导致的光量子效率低,氧化能力低以及光生空穴迁移率低等问题。同时g-c3n4带隙为2.7ev,虽然具有很好的可见光响应性,但是价带位于+1.4v(vs.ag/agcl,ph=6.6),难以氧化水,严重限制了g-c3n4的发展和应用。
随着研究的深入,现在已经有很多学者采用了多种方法来对g-c3n4进行改性,如:与贵金属材料进行复合、与其他半导体复合等,虽然贵金属材料的加入使g-c3n4展现出较为良好的光催化性能,但同时也大大增加了催化剂的成本,限制了g-c3n4的应用,并且不能从根本上改变低的可见光利用率。
技术实现要素:
针对现有技术的以上缺陷或改进需求,本发明提供了一种改性的g-c3n4基可见光光催化剂、其制备方法和应用,其目的在于通过采用nabh4还原的方法对g-c3n4基进行改性,改变其带隙宽度和位置,从而增加其对可见光的响应范围,并通过改变其价带和导带位置,从而使其能光催化氧化水,还原水中溶解氧产过氧化氢,由此解决现有技术对g-c3n4进行改性虽然能够改善其光催化性能但是成本高,且不能从根本上改变其可见光利用率低的技术问题。
为实现上述目的,按照本发明的一个方面,提供了一种改性的g-c3n4基可见光光催化剂,所述光催化剂的直接带隙宽度为2.4~2.7ev、间接带隙宽度为1.7~2.65ev。
优选地,所述光催化剂能够直接吸收波长为550nm的可见光,并能够间接吸收整个可见光区域的可见光。
优选地,所述光催化剂分子结构中包含c≡n,其红外光谱在2180cm-1的位置出现了c≡n的峰。
按照本发明的另一个方面,提供了一种所述的改性的g-c3n4基可见光光催化剂的制备方法,包括如下步骤:
(1)将物质a在400℃~500℃条件下烧制3~5个小时,水洗后得到前驱体g-c3n4;所述物质a在400℃~500℃聚合后能产生三均三嗪结构的g-c3n4;
(2)将步骤(1)所得的前驱体g-c3n4与还原剂混合,在惰性气氛下还原,水洗后即可获得所述的改性的g-c3n4基可见光光催化剂。
优选地,所述物质a为三聚氰胺或尿素。
优选地,所述物质a为三聚氰胺。
优选地,步骤(1)烧制三聚氰胺时升温速率控制在3~15℃/min。
优选地,步骤(2)所述还原剂为nabh4或kbh4。
优选地,步骤(2)所述还原剂为nabh4。
优选地,步骤(2)所述前驱体g-c3n4与所述nabh4混合的质量比为1:(2~6)。
优选地,步骤(2)所述还原温度为300℃~400℃。
优选地,步骤(2)所述还原温度为350℃~370℃。
优选地,步骤(2)所述还原温度为370℃。
优选地,步骤(2)所述还原时间为20min~40min。
优选地,步骤(2)所述还原时间为20min~30min。
优选地,步骤(2)所述还原时间为30min。
优选地,步骤(2)所述水洗次数为2~5次。
按照本发明的另一个方面,提供了一种所述的光催化剂的应用,应用于光催化产过氧化氢。
总体而言,通过本发明所构思的以上技术方案与现有技术相比,能够取得下列有益效果。
(1)现有技术的g-c3n4是一种三均三嗪结构的催化剂,这种结构有利于内过氧键的形成,促进双电子还原的发生,因此对于还原氧气产过氧化氢有一定的特异性。但由于现有技术的g-c3n4价带氧化能力有限,不能氧化水,因此在反应体系为水的情况下,一般不能进行光催化产过氧化氢。本发明是通过nabh4还原的方法,改变g-c3n4的带隙宽度和位置,一方面增加了催化剂对可见光响应范围,利于对可见光的吸收和利用,另外一方面改变了g-c3n4的价带和导带位置,使其能光催化氧化水,还原水中溶解氧产过氧化氢。
(2)本发明在制备g-c3n4前驱体时,选择制备温度为400℃~500℃,优选450℃,而非目前普遍使用的550℃烧制的g-c3n4,不但大大提高了还原后产过氧化氢的能力,而且节省了制备过程中的成本。本发明中改性的g-c3n4基可见光光催化剂的制备不仅需要将g-c3n4前驱体进行还原,而且对g-c3n4前驱体的制备工艺也有要求,本发明中改性的g-c3n4可见光光催化剂性能优良,过氧化氢产量大,是前驱体烧制工艺和还原工艺两个步骤协同配合,共同起作用的结果。
(3)本发明在制备g-c3n4可见光光催化剂时不掺入贵金属,同时使用的材料价格低廉,因此本发明中的g-c3n4光催化剂成本很低。另外本发明中的催化剂制备方法简单,耗时短,因此适用于实际生产和应用。
(4)本发明在制备催化剂的过程中,通过还原改变了g-c3n4的结构。结构的改变有利于电子和空穴的分离,大大降低了电子和空穴重新复合的机率,一方面提高了对可见光的利用效率,另一方面提高了光催化中过氧化氢的产量。
(5)本发明所制备的g-c3n4基光催化剂仍然是一种有机半导体,性质稳定,安全无毒,无重金属,因此可以广泛使用,同时催化剂的制备条件为370℃,在常温下非常稳定。
(6)本发明中的g-c3n4基光催化剂的吸收带宽接近550nm,因此可以吸收可见光,直接利用太阳能进行光催化,在纯水的体系,催化产过氧化氢,实现了储能,这对于可持续发展具有重要意义。
(7)过氧乙酸对于杀菌消毒有非常好的效果,但是由于过氧乙酸的易爆性,十分危险,不便于储存和运输,同时过氧化氢又易于分解,这些因素都限制了过氧乙酸的广泛应用,本发明可以实现以乙酸为反应体系光催化产过氧化氢,进而反应产生过氧乙酸,达到了现配现用的效果,因此本发明有非常广阔的应用前景。
(8)本发明中的g-c3n4可见光光催化剂在实际应用时,可以先负载于多孔材料表面,再进行光催化,一方面可以很好的实现催化剂与反应体系的分离,另外一方面可以减少催化剂的流失,提高催化剂的重复利用率。
附图说明
图1为实施例1g-c3n4前驱体和实施例1还原改性的g-c3n4的uv-vis吸收光谱图;
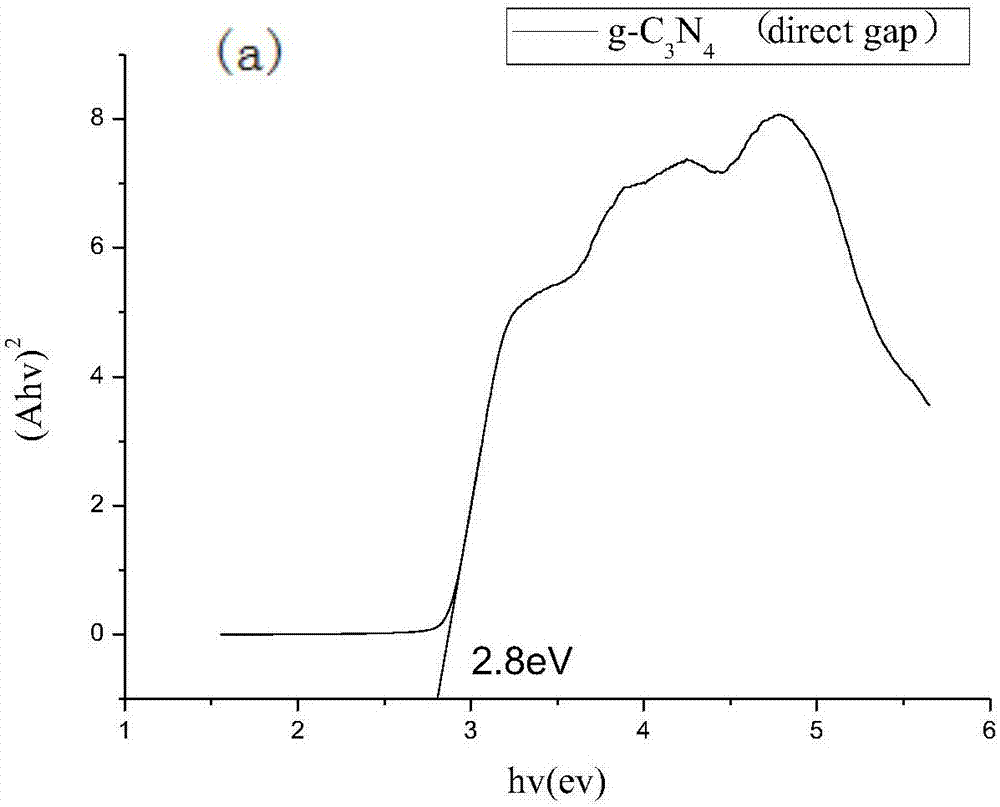
图2为实施例1g-c3n4前驱体和实施例1还原改性的还原g-c3n4的带隙图;
图3为实施例1g-c3n4前驱体和实施例1还原改性的g-c3n4的光致发光光谱;
图4为实施例1g-c3n4前驱体和实施例1还原改性的g-c3n4光照1h体系中过氧化氢产量比较;
图5为实施例1g-c3n4前驱体和实施例1还原改性的g-c3n4的扫描电子显微镜图;
图6为实施例1g-c3n4前驱体和实施例1还原改性的g-c3n4的红外图谱;
图7为对比例1以550℃烧制的g-c3n4为前驱体经过还原制得的g-c3n4和实施例1还原改性的g-c3n4光照1h体系中过氧化氢产量比较。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
以下为实施例:
实施例1
一种改性的g-c3n4基可见光光催化剂的制备方法,包括如下步骤:该光催化剂为可见光响应型g-c3n4基光催化剂。
(1)前驱体的制备
将三聚氰胺放入马弗炉中,升温速率3℃/min,450℃条件下烧4h,使三聚氰胺高温聚合,形成带隙较宽的三均三嗪结构的半导体材料g-c3n4,再水洗干燥去除体系中的其他物质。
(2)制备还原材料
将(1)中所得的材料与nabh4按照质量比为1:5混合研磨后放入管式炉中,370℃氮气条件下还原30min,再将还原后的材料水洗3次,干燥后即可制得改性的g-c3n4基光催化剂。
对本实施例制备的光催化剂进行结果分析:以下提到的g-c3n4是指实施例1步骤(1)在450℃烧制得到的g-c3n4前驱体;还原g-c3n4是指实施例1步骤(1)在450℃烧制得到的g-c3n4前驱体经步骤(2)还原得到的改性的g-c3n4基光催化剂。
(1)g-c3n4和本实施例制备的还原g-c3n4的uv-vis光谱图
如图1所示,从uv-vis光谱图中可以看出450℃烧制的g-c3n4对于可见光的吸收能力明显低于还原g-c3n4。从对可见光吸收的范围来进行比较,450℃烧制的g-c3n4仅能吸收波长小于450nm的光,而本实施例还原g-c3n4能吸收的波长为550nm,间接吸收波长扩展了催化剂的吸收到整个可见光区域,因此还原g-c3n4对于可见光的利用率非常高。
(2)g-c3n4和本实施例的还原g-c3n4的带隙图
从图2中可以看出,传统的g-c3n4只有直接带隙,带隙为2.8ev,如图2(a)所示;而还原后的g-c3n4不仅有直接带隙,而且出现了间接带隙,直接带隙为2.65ev,间接带隙为2ev,分别如图2(b)和图2(c)所示;说明本发明通过对材料的改进,一方面改变了其带隙宽度,增加了对可见光的吸收,另一方面,由于间接带隙的出现,使可见光的吸收范围更广,可见光利用率更高。
(3)g-c3n4和本实施例的还原g-c3n4的光致发光光谱
从光致发光光谱图,图3中可以很明显的看出,450℃烧制的g-c3n4发光强度很高,说明其电子和空穴复合率很高,光催化反应时真正发挥作用的电子和空穴很少。而还原g-c3n4发光强度很弱,说明其电子和空穴的复合效率很低,光催化效率很高,更有利于对可见光的利用。
(4)g-c3n4和本实施例的还原g-c3n4光照1h体系中过氧化氢的产量比较
图4中所示的是450℃烧制的g-c3n4和还原g-c3n4产过氧化氢的比较,反应体系为:100ml去离子水加入到250ml烧杯中,再称取0.1g催化剂,黑暗条件下搅拌2小时后,用300w氙灯照射,每隔半小时取样测过氧化氢浓度。从图中可以很明显看出,450℃烧制的g-c3n4产过氧化氢量很低,接近于0,而经过本发明改进之后催化1h过氧化氢浓度达到了170μmol/l,而且此反应是在未通气体的空气条件下进行,说明本发明有非常好的实用性。
(5)图5g-c3n4(a、c)和本实施例的还原g-c3n4(b、d)的扫描电子显微镜图
从实施例1制得的g-c3n4和还原g-c3n4的扫描电子显微镜图图5可以看出,450℃烧制的g-c3n4是具有粗糙褶皱表面的层状结构,同时表面有些部位呈现圆柱状,而g-c3n4还原之后,层状结构出现断裂,变小,圆柱状的g-c3n4消失,说明还原之后,g-c3n4的形貌发生了变化。
(6)g-c3n4和本实施例的还原g-c3n4的红外图谱
通过对g-c3n4和还原g-c3n4的红外图谱的对比分析,如图6所示,可以明显的发现,还原之后的g-c3n4在2180cm-1的位置出现了新峰,这个位置是c≡n的峰,说明还原之后,产生了c≡n。而c≡n形成后使g-c3n4电子云密度的分布发生了改变,使其价带和导带同时下移。导带下移到‐0.9v(vs.ag/agcl,ph=6.6)左右,仍然有还原氧气产过氧化氢的能力,价带下移到+1.8v(vs.ag/agcl,ph=6.6)左右,具有了氧化水的能力。
通过上述分析可知,本实施例的方法制备的具有可见光催化活性的g-c3n4基光催化剂具有很强的光催化活性和非常高的产过氧化氢的能力,一方面过氧化氢本身无毒,有着非常广泛的应用,另外一方面,通过过氧化氢可以制备出过氧乙酸,过氧乙酸具有很强杀菌效果。本发明可以实现以乙酸为反应体系光催化产过氧化氢,进而反应产生过氧乙酸,实现过氧乙酸的现配现用。
实施例2
一种改性的g-c3n4基可见光光催化剂的制备方法,包括如下步骤:该光催化剂为可见光响应型g-c3n4基光催化剂。
(1)前驱体的制备
将三聚氰胺放入马弗炉中,升温速率15℃/min,400℃条件下烧4h,使三聚氰胺高温聚合,形成带隙较宽的半导体材料g-c3n4,再水洗干燥去除体系中的其他物质。
(2)制备还原材料
将(1)中所得的材料与nabh4按照质量比为1:2混合研磨后放入管式炉中,400℃氮气条件下还原40min,再将还原后的材料水洗3次,干燥后即可制得改性的g-c3n4基光催化剂。
实施例3
一种改性的g-c3n4基可见光光催化剂的制备方法,包括如下步骤:该光催化剂为可见光响应型g-c3n4基光催化剂。
(1)前驱体的制备
将三聚氰胺放入马弗炉中,升温速率5℃/min,500℃条件下烧4h,使三聚氰胺高温聚合,形成带隙较宽的半导体材料g-c3n4,再水洗干燥去除体系中的其他物质。
(2)制备还原材料
将(1)中所得的材料与nabh4按照质量比为1:6混合研磨后放入管式炉中,300℃氮气条件下还原20min,再将还原后的材料水洗3次,干燥后即可制得改性的g-c3n4基光催化剂。
对比例1
现有技术的以550℃烧制的g-c3n4为前驱体还原后制得的g-c3n4制备方法,包括如下步骤:
(1)前驱体的制备
将三聚氰胺放入马弗炉中,升温速率3℃/min,550℃条件下烧4h,使三聚氰胺高温聚合,形成带隙较宽的三均三嗪结构的半导体材料g-c3n4,再水洗干燥去除体系中的其他物质。
(2)制备还原材料
将(1)中所得的材料与nabh4按照质量比为1:5混合研磨后放入管式炉中,370℃氮气条件下还原30min,再将还原后的材料水洗3次,干燥后即可制得该条件下改性的g-c3n4基光催化剂。
将对比例1以550℃烧制的g-c3n4为前驱体还原后制得的g-c3n4和实施例1的还原g-c3n4光照1h体系中过氧化氢的产量比较。传统的高温烧制g-c3n4前驱体的工艺,比如在550℃烧制得到的g-c3n4前驱体,然后同样采用本发明实施例1步骤(2)的还原方法对该前驱体进行还原,得到550℃烧制得到的g-c3n4前驱体经还原后的g-c3n4基光催化剂,并将其与本发明的实施例1在450℃烧制得到的g-c3n4前驱体经步骤(2)还原得到的改性的g-c3n4基光催化剂进行比较。
用目前普遍制备的550℃烧制的g-c3n4为前驱体经过还原后制得的催化剂,即按照对比例1的方法制得的g-c3n4基光催化剂,按照实施例1相同的方法测试过氧化氢产气量,反应体系为:100ml去离子水加入到250ml烧杯中,再称取0.1g催化剂,黑暗条件下搅拌2小时后,用300w氙灯照射,每隔半小时取样测过氧化氢浓度。
从图7中可以看出,在550℃条件下烧制的g-c3n4为前驱体再经过还原,还原后虽然价带和导带位置也有改变,但是由于该温度下烧制使得价带和导带位置仍然不能达到合适的位置,还原后价带仍然无法氧化水,所以在纯水体系中过氧化氢产量依然很低,光催化1h浓度只达到23μmol/l,而实施例1制得的低温g-c3n4经过还原后,光催化1h浓度达到175μmol/l,效果提升了接近9倍。由此也体现出本发明步骤(1)在低温400~500℃烧制g‐c3n4前驱体的必要性,说明本发明的改性的g-c3n4可见光光催化剂的制备不仅需要将g‐c3n4前驱体进行还原,而且对g‐c3n4前驱体的制备工艺也有要求,本发明的改性的g-c3n4可见光光催化剂性能优良,产过氧化氢量大,是前驱体烧制工艺和还原工艺两个步骤协同配合,共同起作用的结果。
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!