用于有机污染物的催化氧化的双金属复合催化剂的制作方法
本发明涉及一种包括金属氧化物基质和金属纳米颗粒沉积物的催化复合材料,以及所述材料用于温和反应条件下有机材料的催化湿式空气氧化的用途。
背景技术:
在本说明书中对先前出版的文件的列出或讨论不必被认为是该文件是现有技术的一部分或是公知常识。
环境污染是当今世界面临的主要关切和挑战之一。在水污染的情况下,从石化、纺织、制药、化学和汽车工业产生大量的工业废水和挥发性有机污染物,所有这些都可能危害环境。因此,在将这些工业废水和废物流排放到周围环境中之前,必须对其进行适当处理,以去除各种有毒、难降解和/或挥发性有机化合物。另外,国家和/或地方政府已经执行越来越严格的法规,仅允许在工业废物和其他废物流排放物中存在少量有机污染物,这不可避免地激发了人们对废水处理的兴趣。
一类最受关注的化合物是有机染料,它们代表了一大类难降解有机污染物。据估计,2003年全球工业使用了10,000多种不同的染料和颜料,并生产了7x105吨以上的合成染料(h.zollinger,彩色化学:有机染料和颜料的合成、性质和应用(colorchemistry:synthesis,propertiesandapplicationsoforganicdyesandpigments),第3修订版,wiley-vch,weinheim,瑞士苏黎世(zurichswitzerland),(2003)637)。在应用过程中,全球染料总产量的约1-20%排放到水生环境中,这可能会导致严重损害水生植物的光合作用活性和人类健康(http://www.food-info.net/uk/colour/azo.htm;m.vakili,等人,carbohydr.polym.,2014,113,115-130;m.yuan,等人,appl.surf.sci.,2011,257,7913-7919)。
迄今为止,已经开发了许多处理废水的工艺。这些工艺包括生物工艺、热分解、吸附、膜工艺和高级氧化工艺(aop)。但是,这样的工艺有其自身的局限性。例如,生物工艺不适用于不可生物降解的化合物和剧毒的化合物的处理,而热分解通常仅用于高化学需氧量(cod)工业废水(典型的cod>10gl-1),其由在高于1000℃的温度的全部燃烧组成。此外,热分解是一个非常耗能的工艺,其产生灰烬,固体废物和含有nox、sox和二噁英的有毒烟雾。吸附和膜工艺非常高效,但需要进行一系列的预处理和后处理。尽管常规的化学氧化方法可以在室温进行,但是需要使用昂贵的且对环境不利的氧化剂,例如氯、二氧化氯或高锰酸钾。大多数aop依赖于使用由紫外线活化的氧(光催化工艺)、臭氧(臭氧化工艺)或过氧化氢(湿式过氧化物氧化和fenton工艺)产生的高活性自由基。然而,用aop例如臭氧化工艺和fenton工艺处理工业废水的成本与cod成比例增加,并且在高cod可变得成本高昂的。
催化湿式空气氧化(cwao)工艺是一种aop,最近由于它以相对低成本使用空气作为氧化剂而备受关注。然而,大多数报道的cwao工艺需要高的反应温度和压力(80-180℃和1-5mpa),这被认为是不希望的(l.mingming,等人,recentpat.chem.eng.,2013,6,79-86)。这些苛刻的反应条件不仅增加了资金投入和运营成本,而且还引起催化剂的严重浸出,这可导致催化剂失活并进一步污染处理后的废水。
最近,xu及其同事报道了在室温和大气压,在mo-zn-al-o催化剂上通过cwao降解红色gtl(y.xu,等人,environ.sci.technol.,2012,46,2856-2863)。张和其同事报道了更活性的催化剂系统,所述催化剂系统使用na2mo4o13/α-moo3复合催化剂在30℃和大气压下经由cwao来降解红色gtl(z.zhang,等人,sci.rep.,2014,4,6797-6805)。尽管这些催化剂具有较高的催化活性,但它们具有浸出问题,并且似乎仅对几种染料起作用,这因此限制了它们的应用。
鉴于以上所述,仍然需要用于通过cwao降解有机污染物的更活性的和稳定的催化剂。更重要的是,这些催化剂必须能够在温和的或环境反应条件下表现出高催化活性。另外,催化剂应具有最小的浸出问题或没有浸出问题,使得它们具有长的使用寿命并且可以被重复使用多次。优选地,所需的催化剂应适合用于气候和自然条件有显著差异的区域(例如,在白天和晚上的温度之间和/或不同季节之间的温度差异都很大的地方),或在偏远的以及欠发达的地区。
技术实现要素:
在本发明的第一方面,提供了一种催化复合材料,其包括:
包括na2mo4o13和α-moo3的混合物的金属氧化物基质,其中所述基质为纳米纤维的形式;和
元素金属纳米颗粒的沉积物,其中各元素金属纳米颗粒由金或钯组成或为其合金,其中
所述元素金属纳米颗粒以相对于所述金属氧化物基质的总重量从0.1至1.2wt%的总量沉积;并且
在所述复合材料中元素金与元素钯的重量:重量比为从1:3至3:1。
在本发明的第一方面的实施方式中:
(ia)元素金属纳米颗粒可以以相对于所述金属氧化物基质的总重量从0.5至1.1wt%的总量沉积,例如从0.75至1wt%;
(ib)在所述复合材料中元素金与元素钯的重量:重量比可以为从0.5:1至2:1,例如1:1;
(ic)所述金属氧化物基质纳米纤维可以具有从500至5,000nm的长度以及从50至500nm的直径,例如从800至4,000nm的长度以及从75至400nm的直径,例如所述金属氧化物基质纳米纤维可以具有从900至3,700nm的长度以及从100至350nm的直径;
(id)在所述金属氧化物基质中na2mo4o13与α-moo3的重量与重量比可以为从0.3:1至0.7:1,例如从0.35:1至0.6:1;
(ie)所述元素金属纳米颗粒的直径可以为从1至50nm,例如从5至30nm,例如从7至25nm。
在本发明的第二方面,提供了如本发明的第一方面所述的催化复合材料及其实施方式的任何技术上合理的组合在有机材料的催化湿式空气氧化中的用途。在本发明的第二方面的实施方式中,所述有机材料可以是选自药物化合物、杀虫剂以及更具体地有机染料(例如番红o、亚甲基蓝和亮绿的一种或多种,或其他具有类似结构的有机化合物)的组的一种或多种。
在本发明的第三方面,提供了一种有机材料的催化湿式空气氧化的方法,所述方法包括以下步骤:在供有包括氧气的气体流的水性环境中使有机材料与本发明的第一方面所述的催化复合材料及其实施方式的任何技术上合理的组合接触以氧化所述有机材料。
在本发明的第三方面的实施方式中:
(ai)所述有机材料可以是选自药物化合物、杀虫剂以及更具体地有机染料(例如番红o、亚甲基蓝和亮绿的一种或多种,或其他具有类似结构的有机污染物)的组的一种或多种;
(aii)所述催化复合材料与有机材料的重量:重量比可以为从1x10-4:1至10:1,例如从1x10-3:1至5:1,例如从0.1:1至1:1;
(aiii)所述方法可以在从0至75℃,例如从2.5至45℃,例如从5至30℃,例如25℃的温度下进行。
在本发明的第四方面,提供了一种形成催化复合材料的方法,所述方法包括以下步骤:
(a)提供包括na2mo4o13和α-moo3的混合物的金属氧化物基质,其中所述基质为纳米纤维的形式;
(b)将所述基质悬浮在溶剂中,并然后添加元素金属源以形成第一混合物;
(c)将配体和还原剂添加到所述第一混合物中并老化持续第一段时间以形成配体涂覆的产物;
(d)收集所述配体涂覆的产物,将其干燥并使其在250至550℃的煅烧温度持续第二段时间,以形成所述催化复合材料,其中:
所述元素金属源由元素金源和元素钯源组成;
所述元素金属源以相对于所述金属氧化物基质的总重量从0.1至1.2wt%的总量将元素金属纳米颗粒沉积在所述基质的表面上;并且
在所述复合材料中元素金与元素钯的重量:重量比为从1:3至3:1。
在本发明的第四方面的实施方式中:
(bi)元素金属纳米颗粒可以以相对于所述金属氧化物基质的总重量从0.5至1.1wt%,例如从0.75至1wt%的总量沉积;
(bii)在所述复合材料中元素金与元素钯的重量:重量比可以为从0.5:1至2:1,例如1:1;
(biii)所述金属氧化物基质纳米纤维可以具有从3,000至35,000nm的长度以及从800至3,100nm的直径,例如从3,500至26,000nm的长度以及从825至2,700nm的直径,例如,所述金属氧化物基质纳米纤维可以具有从3,750至18,000nm的长度以及从850至2,500nm的直径;
(biv)在所述金属氧化物基质中na2mo4o13与α-moo3的重量与重量比可以为从0.3:1至0.7:1,例如从0.35:1至0.6:1;
(bv)所述元素金属纳米颗粒的直径可以为从1至50nm,例如从5至30nm,例如从7至25nm;
(bvi)在用于步骤(a)之前,使所述金属氧化物基质经受从250至400℃的煅烧温度,例如300℃。
(bvii)使所述配体涂覆的产物经受从300至450℃的煅烧温度持续第二段时间以形成所述催化复合材料。
附图说明
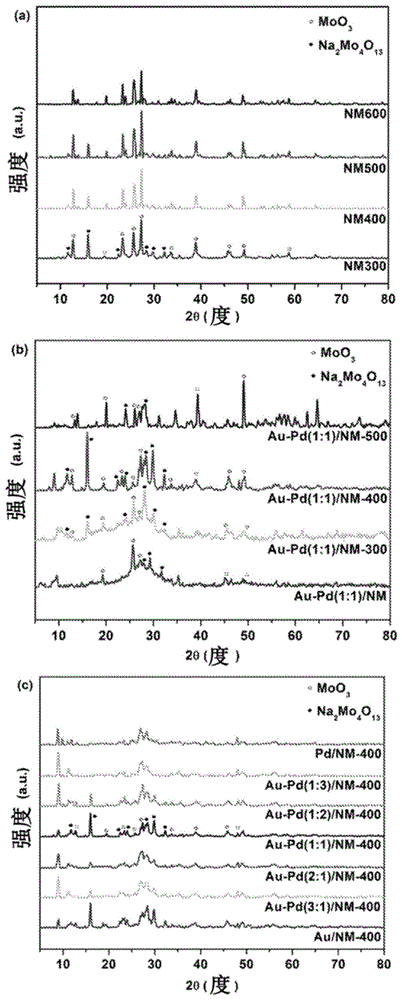
图1描述了以下的xrd图谱:(a)在不同温度煅烧的nm基质;(b)与未经煅烧的样品相比,在不同温度煅烧的本发明的au-pd(1:1)/nm催化剂;以及(c)在400℃煅烧的,具有不同au:pd比的,本发明的au-pd/nm催化剂。
图2描述了以下的fesem图像:(a-b)nm300;以及(c-d)本发明的au-pd(1:1)/nm-400。
图3描述了在初始研究中获得的au-pd(1:1)/nm-400的(a)tem和(b)hrtem图像;(c)优化条件后获得的au-pd(1:1)/nm-400的hrtem图像(其对应于图4(e)中插图的tem图像);以及(d)nm300、au/nm-400、pd/nm-400和au-pd(1:1)/nm-400的drs光谱。
图4描述了本发明的au-pd(1:1)/nm-400的tem、fesem图像和edx元素映射:(a)在初始研究中获得的au-pd(1:1)/nm-400的tem图像;(b-d)对应于(a)中的样品的pd、au和mo的元素映射;(e)优化条件后获得的au-pd(1:1)/nm-400的fesem图像,插图显示了样品的tem图像;以及(f)mo、o、au和pd的元素映射,对应于(e)中插图的tem图像。
图5描述了本发明的nm300和au-pd(1:1)/nm-400的xps光谱,其结合能对应于(a)mo3d;(b)o1s;(c)au4f;和(d)pd3d。
图6描述了煅烧温度对催化剂载体(nm基质)的催化活性的影响,其中(a)和(b)分别代表初始结果和优化结果。在23℃、大气压下,以100mgl-1的初始染料浓度,0.5gl-1的催化剂浓度进行所述实验持续2h的反应时间。
图7描述了煅烧温度对本发明的au-pd(1:1)/nm催化剂的催化活性的影响,其中(a)和(b)分别代表初始结果和优化结果。在23℃、大气压下,以100mgl-1的初始染料浓度,0.5gl-1的催化剂浓度进行所述实验持续2h的反应时间。
图8描述了与nm300相比,使用本发明的au-pd(1:1)/nm-400的染料的颜色变化。在大气压下,在23℃,以100mgl-1的初始染料浓度,0.5gl-1的催化剂浓度进行所述实验。
图9描述了au-pd质量比对本发明的au-pd/nm催化剂的催化活性的影响,其中(a)和(b)分别代表初始结果和优化结果。在大气压下,在23℃,以100mgl-1的初始染料浓度,0.5gl-1的催化剂浓度进行所述实验持续2h的反应时间。
图10描述了au-pd(1:1)/nm-400浓度对染料去除效率的影响,其中(a)和(b)分别代表初始结果和优化结果。在大气压下,在23℃,以100mgl-1的初始染料浓度,分别以0.1gl-1、0.2gl-1以及0.5gl-1的催化剂浓度进行所述实验持续2h的反应时间。对于催化剂浓度为1.0gl-1的实验,反应时间为1h。
图11描述了:(a-c)分别在300mgl-1、400mgl-1和600mgl-1的染料的存在下,对本发明的nm300、au-pd(1:1)/nm-400和au-pd(1:1)nm-500的催化活性的初始研究;以及(d-f)分别在300mgl-1、400mgl-1和600mgl-1的染料的存在下,对本发明的nm300和au-pd(1:1)/nm-400的催化活性的优化研究。在大气压下,在23℃,以0.5gl-1的催化剂浓度进行所述实验持续2h的反应时间。
图12描述了使用本发明的nm300和au-pd(1:1)/nm-400,各种浓度的bg随时间的脱色。在大气压下,在23℃,以0.5g/l的催化剂浓度进行所述实验持续2小时的反应时间。
图13描述了本发明的nm300和au-pd(1:1)/nm-400分别在35℃和45℃的反应温度在去除染料中的催化活性:(a和b)分别在35℃和45℃进行的初始研究;以及(c和d)分别在35℃和45℃进行的优化研究。在大气压下,以600mgl-1的初始染料浓度,0.5gl-1的催化剂浓度进行所述实验持续2h的反应时间。
图14描述了本发明的nm300和au-pd(1:1)/nm-400在5℃和10℃的反应温度在去除染料中的催化活性:(a和b)在5℃,分别进行2h和24h的初始研究;(c和d)在10℃,分别进行2h和24h的初始研究;以及(e-g)在5℃,分别进行2h、24h和48h的优化研究。初始研究(a-d)是以100mgl-1的初始染料浓度进行的,而优化研究(e-g)是以300mgl-1的初始染料浓度进行的。所有的实验是在大气压下以0.5gl-1的催化剂浓度进行的。
图15描述了au-pd(1:1)/nm-400在各种溶液中再生后,在降解:(a)so;(b)mb;以及(c)bg中的可重复使用性和稳定性测试。在大气压下,在23℃,以100mgl-1的初始染料浓度,0.5gl-1的催化剂用量进行所述实验持续2h。
图16描述了:(a)与金属负载量为1%的au-pd(1:1)/nm-400相比,au-pd(1:1)/nm-400(2%)的染料去除效率。在大气压下,在23℃,以100mgl-1的初始染料浓度,0.5gl-1的催化剂用量进行所述实验持续2h;以及(b)au-pd(1:1)/nm-400(2%)样品的tem图像。
图17描述了本发明的nm300、au/nm-400、pd/nm-400以及au-pd(1:1)/nm-400的so降解动力学:(a)在不同的反应时间用各自的催化剂处理过的so样品的uv-vis光谱;(b)c/c0作为反应时间的函数图,其中c表示每个时间点处的染料浓度,以及c0表示初始染料浓度。在大气压下,在23℃,以20mgl-1的初始染料浓度,0.1gl-1的催化剂浓度进行所述实验。
图18描述了本发明的nm300、au/nm-400、pd/nm-400以及au-pd(1:1)/nm-400的mb降解动力学:(a)在不同的反应时间用各自的催化剂处理过的mb样品的uv-vis光谱;(b)c/c0作为反应时间的函数图,其中c表示每个时间点处的染料浓度,以及c0表示初始染料浓度。在大气压下,在23℃,以20mgl-1的初始染料浓度,0.1gl-1的催化剂浓度进行所述实验。
图19描述了本发明的nm300、au/nm-400、pd/nm-400以及au-pd(1:1)/nm-400的bg降解动力学:(a)在不同的反应时间用各自的催化剂处理过的bg样品的uv-vis光谱;(b)c/c0作为反应时间的函数图,其中c表示每个时间点处的染料浓度,以及c0表示初始染料浓度。在大气压下,在23℃,以20mgl-1的初始染料浓度,0.1gl-1的催化剂浓度进行所述实验。
图20描述了所提出的通过使用au-pd(1:1)/nm-400进行染料降解的催化机理:在淬灭剂存在下,(a)so、(b)mb以及(c)bg的降解。在大气压下,在23℃,以20mgl-1的初始染料浓度,0.1gl-1的催化剂浓度进行所述实验;(d)所提出的双金属复合催化剂上染料降解机理的示意图。
图21描述了使用以下将双金属复合催化剂(40)引入废水处理系统的示意图:(a)催化剂的悬浮液;以及(b)在多孔载体(100)上不动的催化剂。
具体实施方式
令人惊讶地发现,可以通过将少量的两种贵金属(金和钯)作为纳米颗粒添加到催化体系的表面来获得具有改善的活性的催化剂。因此,提供了一种催化复合材料,其包括:
包括na2mo4o13和α-moo3的混合物的金属氧化物基质,其中所述基质为纳米纤维的形式;和
元素金属纳米颗粒的沉积物,其中各元素金属纳米颗粒由金或钯组成或为其合金,其中
所述元素金属纳米颗粒以相对于所述金属氧化物基质的总重量从0.1至1.2wt%的总量沉积;并且
在所述复合材料中元素金与元素钯的重量:重量比为从1:3至3:1。
令人惊讶地,相对于基质材料的总重量,包含仅0.1至1.2wt%的元素金属提供了惊人的催化活性。相反,常规系统使用较高重量百分比的金属(例如2至3wt%),但是本文显示,在当前系统中使用如此高的金属负载量导致催化活性的显著损失(参见实施例部分)。
在本文的实施方式中,“包括(comprising)”一词可以被解释为需要所提到的特征,但是不限制其他特征的存在。或者,“包括(comprising)”一词也可以涉及仅意图列出的组分/特征存在的情况(例如,“包括(comprising)”一词可以被短语“由...组成(consistsof)”或“基本上由...组成(consistsessentiallyof)”代替)。明确预期可以将较宽的和较窄的解释都应用于本发明的所有方面和实施方式。换句话说,“包括(comprising)”一词及其同义词可以由短语“由……组成(consistsof)”或短语“基本上由……组成(consistsessentiallyof)”或其同义词代替,反之亦然。
当在本文中使用时,术语“纳米纤维”是指以直径为纳米范围的纤维形式提供的材料。
所述金属氧化物基质由纳米纤维形式的na2mo4o13和α-moo3的混合物组成,其可以通过任何合适的方法获得,例如通过在水热反应条件下使钼氧化物前体(例如(nh4)6mo7o24·4h2o)与合适的钠源(例如nano3)反应,然后煅烧中间体材料。在所述金属氧化物基质中可以使用任何合适的na2mo4o13与α-moo3的重量与重量比。例如,在所述金属氧化物基质中na2mo4o13与α-moo3的重量与重量比可以为从0.3:1至0.7:1或从0.35:1至0.6:1。施加到中间体材料的煅烧温度可以影响所述基质材料中的α-moo3的量。例如,持续5小时的300℃煅烧温度可以提供具有较低的α-moo3比的材料,而持续5小时的600℃煅烧温度的使用可以提供具有高得多的α-moo3比的材料。
如下面更详细地讨论的,金属氧化物基质材料随后在形成以上定义的催化复合材料时经历第二煅烧步骤。该第二煅烧步骤可以进一步改变复合材料的物理特性(即长度和直径)。例如,在用于形成催化复合材料之前,新制备的金属氧化物基质可以具有从3,000至35,000nm的长度以及从800至3,100nm的直径,例如从3,500至26,000nm的长度以及从825至2,700nm的直径,(例如从3,750至18,000nm的长度以及从850至2,500nm的直径)。然而,当材料已经经受了后续的煅烧和/或还原步骤以提供所需的催化复合材料时,纳米纤维的尺寸缩小。例如,所得的金属氧化物基质的纳米纤维可以具有从500至5,000nm的长度以及从50至500nm的直径,例如从800至4,000nm的长度以及从75至400nm的直径,例如所述金属氧化物基质纳米纤维可以具有从900至3,700nm的长度以及从100至350nm的直径。
除非另有明确说明,否则当在本文中使用时,相关范围的端点可以以任何合适的方式组合在一起以形成也被特别考虑的其他范围。例如,在上述新制备的金属氧化物基质中,可以获得以下长度范围:3,000至3,500nm、3,000至3,750nm、3,000至18,000nm、3,000至26,000nm、3,000至35,000nm、3,500至3,750nm、3,500至18,000nm、3,500至26,000nm、3,500至35,000nm、3,750至18,000nm、3,750至26,000nm、3,750至35,000nm、18,000至26,000nm、18,000至35,000nm和26,000至35,000nm。此外,这些长度范围中的任何一个都可以与相应地得出的直径范围中的任何一个结合-这同样适用于此处提到的链接范围的任何分组。
当在本文中使用时,“元素金属”是指金属以元素本身的形式提供。因此,“元素金”是指元素形式的金,其可以是基本上纯的(例如纯度>90%,例如纯度>95%,例如纯度>99.999%),并且可以相应地解释“元素钯”。元素金属纳米颗粒可以基本上由一种金属或另一种金属(即金或钯)形成,但是也可以形成由金和钯的合金形成的纳米颗粒。由金和钯的合金形成的纳米颗粒可以包含任何合适量的金和钯,例如从0.5wt%的金和99.5wt%的钯至99.5wt%的金和0.5wt%的钯。
如上所述,元素金属纳米颗粒可以以相对于金属氧化物基质的总重量从0.1%至1.2wt%的总量沉积。例如,元素金属纳米颗粒可以以相对于金属氧化物基质的总重量从0.5%至1.1wt%的总量沉积。在本发明的特定实施方式中,元素金属纳米颗粒可以以相对于金属氧化物基质的总重量从0.75%至1wt%的总量沉积。如上所述,已经令人惊讶地发现,使用如此少量的金和/或钯的元素金属纳米颗粒在催化性能上提供了显著的改善。
元素金与元素钯的任何合适的重量:重量比都可以用于本文公开的复合材料中。例如,在所述复合材料中元素金与元素钯的重量:重量比可以为从0.5:1至2:1,例如1:1。元素金属纳米颗粒可具有任何合适的纳米颗粒直径。例如,所述元素金属纳米颗粒的直径可以为从1至50nm,例如从5至30nm,例如从7至25nm。
本文公开的催化复合材料适合用于有机材料的分解。例如,药物化合物、杀虫剂以及尤其是有机染料。在某些实施方式中,可分解的有机材料包含至少一个芳香环或芳杂环(例如至少一个苯环)。这样,公开了如上所述的催化复合材料在有机材料的催化湿式空气氧化中的用途。
当在本文中使用时,术语“至少一个芳香环或芳杂环”将被理解为覆盖包含具有4n+2π-电子的环组分的单环系统或稠环系统。每个环或环系统可包含从5个至20个原子,例如从5个至10个原子或6个原子。芳香环系统的实例包括苯和萘。芳杂环的实例包括吡啶和喹啉。在本文中可提及的本发明的特定实施方式中,有机材料可以是包含至少一个芳香环的有机材料,例如苯。
还公开了一种有机材料的催化湿式空气氧化的方法,所述方法包括以下步骤:在供有包括氧气的气体流的水性环境中使有机材料与以上所述的催化复合材料及其实施方式的任何技术上合理的组合接触以氧化所述有机材料。
湿式空气氧化(wao)是一种使用空气中的氧气(或富氧流)以在高的温度(200–325℃)和压力(5–15mpa)下氧化有机物的工艺。在这些严格的反应条件下,有机废物通过自由基机理被分解为中间体(例如羧酸和其他低分子量有机化合物)和最终产物(例如co2和水)。在约250℃下,除乙酸和丙酸外,几乎所有的有机化合物都可以被消除,其中乙酸和丙酸需要约320℃的温度来分解。可以理解,wao需要使用专用设备,从而导致需要在合适的工厂中进行大量的资本投资以及大量的持续的运营成本。催化湿式空气氧化(cwao)通过将温度降低到更适中的水平(通常为125至200℃,最大压力为5mpa)来克服这些问题。理想地,cwao将能够在环境(或低于环境)的温度和压力下运行。
本文所述的用途和方法实现了期望的条件,因为可以在环境压力且以实质上低于wao所需的温度下进行cwao。例如,所述工艺可以在从0至75℃,例如从2.5至45℃,例如从5至30℃,例如25℃的温度下运行。值得注意的是,本文公开的复合材料能够在约0℃或约25℃的温度,在标准压力下,进行cwao工艺,从而使该工艺无需大量的资本投资成本就能操作,并使与该工艺操作相关的持续成本降至最低。
从上面将注意到,上述的用途和方法可以用于氧化任何有机材料。例如,有机材料可以是有机染料-但此工艺不仅限于有机染料。本文可提及的有机染料的实例包括但不限于番红o、亚甲基蓝、亮绿以及其他具有类似结构的有机染料(例如具有至少一个芳香环或芳杂环,例如苯环)。可以通过本文公开的方法氧化的其他有机材料的实例包括但不限于药物化合物和杀虫剂化合物,例如含有至少一个苯环的药物化合物和杀虫剂化合物。
当在本文中使用时,关于cwao方法的术语“水性环境”是指有机材料和催化复合材料在基本上由水组成的溶剂中(例如,水占溶剂的从90至100wt%,例如大于95wt%,大于99wt%,例如大于99.999wt%)。如果存在有机溶剂,则不将其视为溶剂的一部分,而将其视为有机材料的一部分。
本文讨论的cwao方法需要将包括氧气的气体流供应至溶剂、有机材料和催化复合材料的混合物,以实现所需的氧化。该气体可以是包含氧气的任何气体,例如空气或纯氧气(例如来自气瓶)。所述领域的技术人员使用他们的知识和反复试验可以容易地确定所需的气体的量和流量。
可以理解,可以以合适的量使用催化复合材料以进行反应。当在本文中使用时,在一般术语中使用的“催化剂”是指引起或加速化学反应而自身不受到影响的材料。换句话说,催化复合材料可以多次使用,但是可以相对于要氧化的有机材料以亚化学计量的量存在,或者其可以过量存在。例如,催化复合材料与有机材料的重量:重量比可以为从1x10-4:1至10:1,例如从1x10-3:1至5:1,例如从0.1:1至1:1。
可以理解,必须制备催化复合材料以用作催化剂。这可以通过任何合适的途径来实现,例如形成催化复合材料的方法,该方法包括以下步骤:
(a)提供包括na2mo4o13和α-moo3的混合物的金属氧化物基质,其中所述基质为纳米纤维的形式;
(b)将所述基质悬浮在溶剂中,并然后添加元素金属源以形成第一混合物;
(c)将配体和还原剂添加到所述第一混合物中并老化持续第一段时间以形成配体涂覆的产物;
(d)收集所述配体涂覆的产物,将其干燥并使其经受从250至550℃的煅烧温度持续第二段时间,以形成所述催化复合材料,其中:
所述元素金属源由元素金源和元素钯源组成;
所述元素金属源以相对于所述金属氧化物基质的总重量从0.1%至1.2wt%的总量将元素金属纳米颗粒沉积在所述基质的表面上;和
在所述复合材料中元素金与元素钯的重量:重量比为从1:3至3:1。
在本文可提及的某些实施方式中,在步骤(d)中,可使配体涂覆的产物经受从300至450℃的煅烧温度。
在上文和以下实施例部分中讨论了在步骤(a)中提供的金属氧化物基质(即,新鲜制备的金属氧化物材料)的物理性质及其制备。如上所述,可以使用包括第一煅烧步骤的方法来制备以上步骤(a)中提供的金属氧化物基质。在步骤(a)中使用之前,该第一煅烧步骤可以在从250至400℃,例如300℃的煅烧温度下进行。
可以理解,形成本文所述的催化复合材料的组分材料的物理性质和量与上述的那些相同。例如:
(ci)元素金属纳米颗粒可以以相对于金属氧化物基质的总重量从0.5%至1.1wt%,例如从0.75至1wt%的总量沉积;在所述复合材料中元素金与元素钯的重量:重量比可以为从0.5:1至2:1,例如1:1;
(cii)在所述金属氧化物基质中na2mo4o13与α-moo3的重量与重量比可以为从0.3:1至0.7:1,例如从0.35:1至0.6:1;
(ciii)所述元素金属纳米颗粒的直径可以为从1至50nm,例如从5至30nm,例如从7至25nm。
在上述工艺的步骤(b)中,可以使用任何合适的溶剂、任何元素金源和任何元素钯源。合适的溶剂的实例包括但不限于水和相容的有机溶剂,尽管优选单独使用水。合适的元素金源的实例包括但不限于aucl3或更具体地haucl4及其水合物。合适的元素钯源的实例包括但不限于pd(no3)2、h2pdcl4、na2pdcl4,或更具体地pdcl2、pdbr2等。
在上述工艺的步骤(c)中,可将任何合适的配体和还原剂用于形成配体涂覆的产物。配体应该是可以与存在于各自的元素金属源中的金金属离子和钯金属离子形成配合物的配体。合适的配体的实例包括但不限于柠檬酸盐(例如柠檬酸钾或更具体地柠檬酸钠)等。还原剂应该能够将金离子和钯离子还原成它们各自的元素金属。合适的还原剂的实例包括但不限于氢碘酸,或更具体地,硼氢化钠。第一段时间可以是任何合适的时间段,以确保形成配体涂覆的产物(即,沉积在基质上但涂覆有配体的元素金属纳米颗粒)。合适的时间段的实例包括但不限于从30分钟至24小时,例如从45分钟至1.5小时,例如约1小时的时间段。
在步骤(d)中,可以控制煅烧温度以保持煅烧后基质中na2mo4o13的比例与上述的比一致。因此,用于煅烧的合适温度范围可以是从250至550℃,例如从300至450℃。煅烧时间可以是足够的时间段以确保配体涂层的基本去除。合适的时间段的实例包括但不限于,在以上刚刚讨论的煅烧温度下,从30分钟至1.5小时,例如从45分钟至1.25小时,例如约1小时的时间段。
现在将关于以下非限制性实施例讨论本发明的其他方面和实施方式。
实施例
材料
购自sigma-aldrich的钼酸铵四水合物((nh4)6mo7o24·4h2o,99%)、硝酸钠(nano3,99%)、氯化钯(pdcl2,99%)、柠檬酸三钠二水合物(na3c6h5o7·2h2o,99%)、氢氧化钠(naoh,98%)、盐酸(hcl,37%)、丙酮(ch3coch3,99.9%)、1,4-苯醌(bq)、叠氮化钠(nan3,99.5%)、二甲基亚砜(dmso,99.7%)和亮绿(bg)。购自vwr新加坡私人有限公司的四氯金酸三水合物(haucl4·3h2o,99%)、硼氢化钠(nabh4,98%)、番红o(so)和亚甲基蓝(mb)。所有化学品均按原样使用,无需进一步净化。除非另有说明,否则在整个研究中均使用电阻率为18.2mωcm的milliporeco.milliq(mq)水。
一般方法
在brukerd8advancex-射线衍射仪上,在5-80°的2θ范围内,以cukα辐射
催化剂载体基质的合成
原则上,稳定的金属氧化物及其与其他稳定材料的复合材料可用作用于双金属复合催化剂的基质。由于其报道的催化活性,na2mo4o13/α-moo3(nm)被选为用于复合催化剂的实例基质,并因此还可以作为用于比较的基准。nm基质是使用从报道的方法(z.zhang,等人,sci.rep.2014,4,6797-6805)改进的水热方法来合成的。
通常,将5.296g(nh4)6mo7o24·4h2o溶解在30mlmq水中,以在50℃下形成澄清溶液,然后添加0.408gnano3并在磁力搅拌下混合持续30分钟。随后,将所得溶液转移到50ml衬有聚四氟乙烯的不锈钢高压釜中,并在120℃下进行水热处理持续18h。通过离心分离产生的沉淀物,用mq水彻底洗涤,然后在60℃下在炉中干燥过夜。将沉淀物研磨成细粉后,将获得的样品在马弗炉中空气气氛下分别于300℃、400℃、500℃和600℃煅烧持续5h,以获得最终产物,其分别表示为nm300、nm400、nm500和nm600。
可以用作催化剂载体的金属氧化物基质的另一实例是femgal混合氧化物催化剂。所述femgal的混合氧化物是通过自动燃烧法来合成的(
评价合成的催化剂的催化活性的通用方法
用三种染料(so、mb和bg)作为目标污染物评估了合成的催化剂的催化活性。在容量为250ml的玻璃反应器中,在环境条件下,以1.2lmin-1的流速将空气泵入悬浮液的底部,研究了染料在合成的催化剂上的降解。
在一个典型的实验中,在mq水中,催化剂浓度为0.5gl-1,并且初始单一原始染料浓度在100至600mgl-1之间。为了染料降解动力学研究,使用了0.1gl-1的催化剂浓度和20mgl-1的初始单一原始染料浓度。在催化期间,以适当的时间间隔收集样品等分试样并过滤以进行分析。使用uv-2600uv-vis分光光度计,以在染料的最大吸收波长(对于bg为625nm,对于so为517nm,以及对于mb为664nm)处测量染料浓度。用shimadzuasi-vtoc分析仪测量总有机碳(toc)中的变化。另外,还通过在添加催化剂之前用n2吹洗染料溶液持续30min并在添加催化剂之后将n2连续注入悬浮液的底部来进行染料在催化剂上的吸附,以防止溶解于h2o中的o2的影响。
为了研究合成的催化剂的可重复使用性,将反应后获得的悬浮液离心,并用mq水;hcl溶液;naoh溶液;或丙酮洗涤三次。收集再生的催化剂,在重复使用前,在105℃下干燥持续2h。
显示煅烧温度对nm基质的催化活性影响的研究
在环境条件(在23℃的室温以及大气压)下评估了在不同的煅烧温度下制备的各种nm基质(nm300、nm400、nm500和nm600)对三种染料(so、mb和bg)的cwao,其中原始染料浓度为100mgl-1以及催化剂浓度为0.5gl-1。初始研究以及后续优化研究的结果(分别显示在图6a和图6b中)表明,与nm400、nm500和nm600相比,nm300对三种染料的降解表现出最高的催化活性。这可能是由于在nm300中α-moo3和na2mo4o13两者的共存。xrd结果(图1a)表明,随着煅烧温度的升高,moo3的量比na2mo4o13更高,这是因为在较高的温度下na2mo4o13向moo3的转化率更高。因此,高比例的moo3/na2mo4o13降低了催化剂的催化性能,这与报道(z.zhang,等人,sci.rep.2014,4,6797-6805)相符。因此,在实施例1中选择nm300作为用于制备双金属复合催化剂的基质载体,并在随后的实施例中作为基准催化剂。
实施例1.通过金属颗粒在na2mo4o13/α-moo3(nm)基质上的沉积来制备双金属复合催化剂
选择湿溶液途径,通过将金属颗粒沉积到nm基质上来制备本发明的双金属复合催化剂。使用简便的一锅沉积还原方法,以nm300为载体,并且以haucl4·3h2o和pdcl2分别为金源和钯源,合成了一系列总金属负载量为1wt%的负载有au-pd的nm催化剂。
通常,在超声处理下,将500mg的nm300分散在10-20mlmq水中持续30min。然后,在磁力搅拌下将520μlhaucl4·3h2o溶液(24.28mm)和2090μlpdcl2溶液(11.28mm)加入上述悬浮液中,并允许将反应混合物搅拌持续10min。随后,在连续搅拌下加入5ml柠檬酸钠溶液(0.05m),并搅拌持续30min。随后在连续搅拌下快速加入5ml新鲜制备的nabh4溶液(0.05m),并将反应混合物搅拌持续60min。通过离心获得催化剂悬浮液,并用mq水洗涤。将获得的样品真空干燥过夜,并然后在马弗炉中空气下分别在300℃、400℃和500℃煅烧持续1h,以获得最终产物,其分别表示为au-pd(1:1)/nm-300、au-pd(1:1)/nm-400和au-pd(1:1)/nm-500。
作为参考,通过调节haucl4·3h2o溶液和pdcl2溶液的添加量,根据上述一锅沉积还原方法,以不同的au-pd质量比(1:3、1:2、2:1、3:1),制备了总金属负载量为1wt%的负载有au-pd的nm催化剂。此外,分别制备具有相同的总金属负载量为1wt%的au/nm和pd/nm。
为清楚起见,在表1总结了已合成的各种金属复合催化剂以及不同的条件。
表1.使用不同条件制备的金属复合催化剂。
实施例2.与na2mo4o13/α-moo3(nm)基质相比,合成的双金属复合催化剂的表征
通过x-射线衍射(xrd)表征
进行xrd分析以表征催化剂的晶体结构。从nm300的图1a可以看出,观察到位于约12.70°、19.38°、23.33°、25.77°、27.28°、33.67°、38.86°、46.07°、49.34°和58.75°的衍射峰并且可以分配给α-moo3(jcpdsno.35-0609),而位于约11.75°、15.96°、22.50°、28.36°、29.72°和32.18°的衍射峰可以分配给na2mo4o13(jcpdsno.28-1112)。这表明nm300是含有α-moo3和na2mo4o13两者的复合材料(z.zhang,等人,sci.rep.,2014,4,6797-6805)。与nm300相比,对于nm400、nm500和nm600,na2mo4o13的衍射峰的强度缓慢降低,这可能是由于随着煅烧温度的增加,na2mo4o13向α-moo3的转化(图1a)。
使用topas软件对xrd光谱中各种nm基质中na2mo4o13与α-moo3的比进行量化(表2)。观察到,随着煅烧温度的增加,更多的na2mo4o13被转化为α-moo3。对于具有au-pd沉积物的催化剂,峰受到au-pd的存在的影响。因此,无法直接量化确切的比例。然而,推论出,该比例应与在相同温度煅烧的相应nm基质在相同范围内。
表2.对于在不同温度煅烧的nm基质,na2mo4o13与α-moo3的比。
此外,在煅烧前后,au-pd(1:1)/nm样品中观察到moo3和na2mo4o13两者的衍射峰(图1b)。对于在400℃煅烧后具有不同的au-pd质量比的催化剂也观察到类似的现象,表明负载有au和pd两者后,没有载体的结构的变化(图1c)。
然而,对于所有负载au-pd的催化剂,对于au和pd两者均未观察到衍射峰,其衍射峰应分别位于约38.2°和44.4°以及位于约40.1°和46.7°(a.cybula,等人,appl.catal.,b:environ.,2014,152,202-211)。这可能是由于催化剂中au和pd两者的高分散度和低负载量。
通过场发射扫描电子显微镜(fesem)、透射电子显微镜x-射线能谱仪(tem-edx)和uv-vis漫反射光谱(drs)进行表征。
通过fesem表征了合成的nm300和au-pd(1:1)/nm-400催化剂。nm300的fesem图像显示了纳米纤维形貌(图2a和图2b),证实了与z.zhang,等人,sci.rep.,2014,4,6797-6805中报道的相同的形貌。此外,au-pd(1:1)/nm-400的fesem图像显示出与nm300相似的纳米纤维形貌,表明au-pd沉积物对催化剂的形貌影响很小(图2c、图2d和图4e)。
此外,在初始研究期间获得的au-pd(1:1)/nm-400样品的tem图像显示,au纳米颗粒和pd纳米颗粒在某种程度上未均匀地沉积在基质上(图3a)。如hrtem图像(图3b)所示,约0.366和0.385nm的晶格条纹之间的间距被分别分配给α–moo3的(100)平面和(001)平面(z.zhang,等人,sci.rep.,2014,4,6797-6805)。此外,0.224nm和0.232nm的条纹间距离可以被分别分配到pd纳米颗粒和au纳米颗粒的(111)平面(w.fang,等人,j.colloidinterfacesci.,2017,490,834-843),且0.228的条纹间距离也在0.224nm至0.232nm的范围内。可以将0.201和0.206nm的条纹间距离分配给au纳米颗粒的(200)平面(r.h.padilla,等人,ultrason.sonochem.,partb,2017,35,631-639)。
在使用最优化条件表征au-pd(1:1)/nm-400的样品中,从图3c的hrtem图像观察到0.231nm的条纹间距离位于各个au(111)平面(0.236nm)和pd(111)平面(0.225nm)的晶格距离之间,这证实了在au-pd(1:1)/nm-400中形成了au-pd合金颗粒(g.zhan,等人,mater.lett.,2011,65,2989-2991;z.zhuang,等人,j.powersources,2015,291,132-137)。
此外,还通过drs分析验证了au-pd(1:1)/nm-400中au-pd合金颗粒的形成(图3d)。在au/nm-400中发现了在约560nm处的最大表面等离子体共振(spr)带。在pd/nm-400中发现了所谓的具有广泛光学共振的局部表面等离子体激元(lsp)。然而,在au-pd(1:1)/nm-400的光谱中未观察到spr峰,这表明pd抑制了au的等离子体波带,可能是由于au和pd之间的强相互作用以及在au-pd(1:1)/nm-400中au-pd合金颗粒的形成(r.h.padilla,等人,ultrason.sonochem.,partb,2017,35,631-639)。
如图4a-d中所示,通过tem-edx元素映射进一步证实了初始研究中产生的au-pd(1:1)/nm-400中au纳米颗粒和pd纳米颗粒的沉积。随着催化剂制备方法的进一步改进,au纳米颗粒和pd纳米颗粒在表面上的均匀沉积得以优化,以产生具有图4e和图4f中所示元素映射的au-pd(1:1)/nm-400样品。由以上xrd结果支持,可以得出结论,已经成功制备了沉积有金属纳米颗粒的催化剂载体。
表3中列出了纳米纤维的长度和宽度以及金属纳米颗粒的直径的值的范围。
表3.用于催化剂的纳米纤维的长度和宽度以及金属纳米颗粒的直径的值的范围。
a具有1wt%的总金属负载量。
b来自fesem的数据。
c来自tem的数据。
通过x-射线光电子能谱(xps)表征
进行xps分析以研究载体和催化剂的表面化学组成和化学基本态。图5a-d示出了在nm300和au-pd(1:1)/nm-400中mo3d、o1s、au4f和pd3d的xps光谱。在nm300的mo3dxps光谱中观察到处于232.2ev和235.4ev的两个峰,而在au-pd(1:1)/nm-400的mo3d光谱中观察到处于232.3ev和235.5ev的峰。这表明在两个催化剂中,mo元素主要以mo6+的化学态存在(w.li,等人,appl.catal.,b:environ.,2009,92,333-340)。
对于如图5b中所示的o1sxps光谱,在nm300中530.1ev处的峰和在au-pd(1:1)/nm-400中529.9ev处的峰归因于金属氧化物的o2-离子(晶格氧)。在nm300中531.7ev处的峰和在au-pd(1:1)/nm-400中531.9ev处的峰被分配给氧空位区域中的o2-离子,而在nm300中534.2ev处的峰和在au-pd(1:1)/nm-400中534.0ev处的峰被分配到松散结合的表面o2(z.zhang,等人,sci.rep.2014,4,6797-6805;j.huang,等人,appl.surf.sci.,2010,257,116-121)。
au-pd(1:1)/nm-400中的松散结合的表面o2和氧空位区域中o2-离子的百分比高于nm300中的百分比(分别为6%相对于4%;以及27%相对于24%)。这些结果表明,au-pd(1:1)沉积后,分子氧的吸附和活化的能力增强,这可导致au-pd(1:1)/nm-400的催化剂活性高于nm300。考虑到该反应是在环境条件进行的,因此催化湿式空气氧化(cwao)反应中的晶格氧参与的可能性应该很小,因此吸附的氧类应该是参与cwao反应的主要活性氧类。
图5c示出了au-pd(1:1)/nm-400的au4fxps光谱。将84.5ev和88.1ev处的峰分配给au0的au4f7/2和au4f5/2,而将86.2ev和89.8ev处的峰分配给au3+的au4f7/2和au4f5/2(w.fang,等人,j.colloidinterfacesci.,2017,490,834-843;g.fu,等人,crystengcomm,2014,16,1606-1610;s.m.kim,等人,acsnano,2011,5,1236-1242)。
在图5d中示出了au-pd(1:1)/nm-400的pd3dxps光谱,并且所述光谱由四个子峰组成。336.3ev和341.6ev处的峰归因于pd0的pd3d5/2和pd3d3/2,而将338.8ev和344.2ev处的峰分配给pd2+(w.fang,等人,j.colloidinterfacesci.,2017,490,834-843)。表面pd2+类的存在可能是由于催化剂在空气中的煅烧。另外,观察到,与块状au金属和pd金属的标准值相比,au0和pd0两者峰的结合能向更高的结合能移动,这表明au和pd两者的电子不足或电子损失(h.zhang,等人,nat.mater.,2011,11,49)。au-pd双金属颗粒的电子似乎在au-pd与moo3之间的界面处被转移到moo3中的相邻o-空位点,并且这些o-空位点可以充当用于分子氧的吸附的位点(h.zhang,等人,nat.mater.,2011,11,49)。
催化剂的brunauer-emmett-teller(bet)表面积
催化剂的比面积非常小,例如对于au-pd(1:1)/nm-400和nm300分别为约5.4和8.5m2/g。在催化评估之前,进行吸附实验,其表明bg在au-pd(1:1)/nm-400和nm300上在30min内的吸附小于10%。这表明染料分子未主要吸附在这些样品上,这支持了催化剂的小比表面积。另外,这也表明在随后的染料降解实验中染料的去除主要是由于催化氧化而不是由于染料的吸附。
实施例3.煅烧温度对au-pd(1:1)/nm催化剂的催化活性的影响
根据实施例1制备在不同温度处煅烧的负载有1:1质量比的au-pd的nm300基质,以研究煅烧温度对染料去除效率的影响。在大气压下,在23℃,以100mgl-1的初始染料浓度,0.5gl-1的催化剂浓度进行所述实验持续2h的反应时间。
初始研究
在初始研究中,如图7a所示,未煅烧的au-pd(1:1)/nm显示可忽略不计的催化活性,以及煅烧后所述催化活性得到了增强,这可能是由于柠檬酸盐配体的烧去。由于au纳米颗粒和pd纳米颗粒的催化功能,与nm300相比,au-pd(1:1)/nm-400催化剂表现出更高的催化活性。
然而,随着煅烧温度的进一步升高,染料去除效率变得稍低。在图7a中注意到,由于nm300和au-pd(1:1)/nm-400的toc去除效率已达到近90%,因此未清晰地观察到au和pd的增强的催化效果。在实施例6和实施例7的初始研究中可以更清晰地观察到这一点,其中使用了较高的染料浓度和较宽的反应温度范围。
随后的优化研究
类似地,如图7b所示的优化结果表明,煅烧后由于柠檬酸盐配体的烧去,au-pd(1:1)/nm的催化活性得到增强。观察到au-pd(1:1)/nm-400催化剂是活性最高的催化剂,因为只有在400℃以上才能完全去除覆盖au纳米颗粒和pd纳米颗粒的配体(z.bai,等人,j.ofpowersources,2010,195,2653-2658)。然而,由于na2mo4o13向moo3的转化率较高,因此在高于400℃的温度下煅烧导致moo3与na2mo4o13的比更高。因此,观察到au-pd(1:1)/nm-500的染料去除效率低于au-pd(1:1)/nm-400的染料去除效率。
此外,由于即使nm300具有较高的na2mo4o13含量,au纳米颗粒和pd纳米颗粒仍具有强的催化功能,所以au-pd(1:1)/nm-400也表现出比nm300更高的催化活性(将图7b中的au-pd(1:1)/nm-400与图6b中的nm300比较)。
如图8所示,从颜色变化可以更清晰地观察到染料的降解。对于nm300和au-pd(1:1)/nm-400催化剂两者,将含有so的溶液在最短的时间内降解,而含有bg的溶液花费三种染料溶液中最长的时间来降解。还清晰地观察到,使用au-pd(1:1)/nm-400,含bg溶液的脱色要比使用nm300快得多(见图6b和图7b)。但是,经过2h的反应时间后,所有三种染料都可以被有效地降解(图7a和图7b)。
实施例4.au-pd质量比对au-pd/nm催化剂的催化活性的影响
根据实施例1制备了一系列在400℃煅烧的负载有不同au-pd质量比(总金属负载量为1wt%)的nm300,并评估了其对于so、mb和bg去除的催化活性。在大气压下,在23℃,以100mgl-1的初始染料浓度,0.5gl-1的催化剂浓度进行所述实验持续2h的反应时间。
图9a和图9b分别显示了催化剂的催化活性的初始结果和优化结果。两项研究均表明,与具有其他au-pd质量比的催化剂以及单金属au/nm-400和单金属pd/nm-400相比,使用au-pd(1:1)/nm-400实现了更有效的so、mb和bg去除。这表明最佳的au-pd比为1:1,并且au和pd两者的双金属组合比仅使用单个au或pd金属更好,这可能是由于au和pd的协同效应。
实施例5.催化剂浓度对au-pd/nm-400催化剂的催化活性的影响
还研究了催化剂浓度对催化剂(根据实施例1制备的)的染料去除效率的影响。在初始研究和优化研究两者中,使用0.1gl-1、0.2gl-1和0.5gl-1的催化剂浓度,并且在大气压下,在23℃,以100mgl-1的初始染料浓度进行所述实验持续2h的反应时间。对于催化剂浓度为1.0gl-1的实验,反应时间为1h。
分别如图10a和图10b所示,观察到染料去除效率随催化剂浓度的增加而增加。反应2h后,使用0.5gl-1au-pd(1:1)/nm-400可以去除90%以上的so、mb和bg。此外,使用1.0gl-1au-pd(1:1)/nm-400,持续1h的反应时间,可以达到类似的去除效率。这可能是由于存在更多具有较高催化剂浓度的催化位点。
实施例6.染料浓度对nm300、au-pd(1:1)/nm-400和au-pd(1:1)/nm-500催化剂的染料去除效率的影响
以固定的催化剂浓度,使用根据实施例1制备的nm300、au-pd(1:1)/nm-400和au-pd(1:1)/nm-500,使用不同的染料浓度,研究催化剂活性。在初始研究和优化研究两者中,使用300mgl-1、400mgl-1和600mgl-1的染料浓度,并且在大气压下,在23℃,以0.5gl-1的催化剂浓度进行所述实验持续2h的反应时间。
初始研究
以固定的0.5gl-1的催化剂用量,以不同的染料浓度,研究催化剂活性。发现在300mgl-1的三种染料浓度,au-pd(1:1)/nm-400表现出比au-pd(1:1)/nm-500和nm300更高的催化活性(图11a)。当染料浓度增加到400mgl-1时,对于三种染料,au-pd(1:1)/nm-400仍显示出高的toc去除(>90%),而当使用nm300时,对于mb和bg,toc去除<80%(图11b)。当染料浓度进一步增加到600mgl-1时,au-pd(1:1)/nm-400的催化活性(so~90%,mb~75%和bg~55%)仍高于nm300的催化活性(so~75%,mb~40%和bg~30%)(图11c)。这些结果表明,au-pd在载体上的沉积可以显著提高高染料浓度处nm300的催化性能。
随后的优化研究
对于随后使用300和400mgl-1的染料的优化研究(图11d和图11e),au-pd(1:1)/nm-400和nm300的so去除效率相似(>90%),而au-pd(1:1)/nm-400的mb和bg去除效率(>90%)比nm300高得多。随着染料浓度增加到600mgl-1,au-pd(1:1)/nm-400催化剂对于这三种染料仍然表现出高的toc去除效率(图11f)。这表明,由于au-pd的沉积,与高的染料浓度的nm300相比,au-pd(1:1)/nm-400具有更高的催化活性。此外,在图12中视觉上观察到,其中对于300mgl-1和400mgl-1的bg浓度,使用au-pd(1:1)/nm-400比使用nm300产生更剧烈的bg的脱色。
实施例7.与nm300相比,反应温度对au-pd(1:1)/nm-400催化活性的影响
分别在5℃、10℃、35℃和45℃的反应温度进行该实验,以评估反应温度对au-pd(1:1)/nm-400(根据实施例1制备的)和nm300的催化活性的影响。
对于在35℃和45℃处进行的实验(初始研究和优化研究两者,图13a-d),在大气压下,以600mgl-1的初始染料浓度,0.5gl-1的催化剂浓度进行所述实验持续2h的反应时间。
对于在5℃和10℃进行的实验,在大气压下,以100mgl-1的初始染料浓度,0.5mgl-1的催化剂浓度进行所述初始研究(图14a-d)持续2h和24h。对于优化研究(图14e-g),在大气压下,在5℃,以300mgl-1的初始染料浓度,0.5mgl-1的催化剂浓度进行所述优化研究持续2h、24h和48h。
初始研究
从初始结果观察到,在35℃和45℃时au-pd(1:1)/nm-400的催化活性通常高于nm300的催化活性(图13a和图13b)。随着反应温度从35℃升高到45℃,染料去除效率增加。
分别在5℃和10℃的反应温度下,以100mgl-1的初始染料浓度进一步研究染料去除效率。图14a-d示出了从5℃到10℃增加的催化活性,并且随着将反应时间从2h延长至24h,染料去除被增强。这表明au-pd(1:1)/nm-400可以在低于室温的温度下工作,并且有潜力用于气候和自然条件有显著差异的地区。例如,在白天和黑夜以及在不同季节中,温度差异都很大的地区。
随后的优化研究
与初始结果相似,在优化结果中观察到,在35℃下,对于去除mb和bg,au-pd(1:1)/nm-400的催化活性高于nm300的催化活性(图13c)。随着运行温度升高到45℃,在nm300上增加的染料去除效率,其与在au-pd(1:1)/nm-400上增加的染料去除效率相似(图13d)。简而言之,这表明运行温度越高,催化活性越高。
考虑到温度随一天中的时间、区域和季节的变化,进一步研究了au-pd(1:1)/nm-400和nm300在5℃的催化活性。如图14e所示,在5℃,以300mgl-1的染料,对于au-pd(1:1)/nm-400,2h后的染料去除效率(对于so、mb和bg分别为61%、52%和45%)高于nm300的染料去除效率(对于so、mb和bg分别为40%、19%和10%)。将反应时间延长至48h后,对于au-pd(1:1)/nm-400,so、mb和bg的染料去除效率分别达到92%以上、88%以上和85%以上,而对于nm300,so、mb和bg的该效率分别为68%、39%和26%(图14g)。这表明即使在5℃的低反应温度,au-pd(1:1)/nm-400仍然有效且能够去除染料。由此看来,au-pd(1:1)/nm-400有潜力用于温度差异显著的区域。
实施例8.au-pd(1:1)/nm-400催化剂的可重复使用性和稳定性
进行失活测试以研究用于染料去除的au-pd(1:1)/nm-400(根据实施例1制备的)的耐久性。图15a-c显示,在催化剂重复使用五次之后,对于三种不同的染料均实现了高的染料去除效率。还观察到催化活性不受用于再生或洗涤催化剂的溶液(水、丙酮、hcl、naoh)的选择的影响,因此其表明了催化剂的一般用途和高稳定性。另外,染料氧化10h后从au-pd(1:1)/nm-400释放的au3+和pd2+的重量百分比分别为3.3%和1.5%。因此可以得出结论au-pd(1:1)/nm-400具有良好的可重复使用性和催化稳定性。
实施例9.金属负载量(重量%)对au-pd(1:1)/nm-400催化剂的催化性能的影响
作为比较研究,根据实施例1中的au-pd(1:1)/nm-400的类似的方法制备负载有2wt%的金属(质量比为1:1的au-pd)的au-pd(1:1)/nm-400(2%)。在大气压下,在23℃,以100mgl-1的初始染料浓度,0.5gl-1的催化剂浓度进行所述催化研究持续2h。
如图16a所示,au-pd(1:1)/nm-400(2%)降解三种不同染料的催化活性低于au-pd(1:1)/nm-400的催化活性,其具有1wt%的金属负载量。这可能是由于2wt%的较高金属负载量处au-pd纳米颗粒的聚集(如图16b的tem图像所示),因此其降低了au-pd(1:1)/nm-400(2%)的催化活性。
实施例10.so、mb和bg在nm300、au/nm-400、pd/nm-400和au-pd(1:1)/nm-400上的降解动力学
为了进一步理解催化剂的催化性能,使用20mgl-1的低染料浓度研究so、mb和bg在nm300、au/nm-400、pd/nm-400和au-pd(1:1)/nm-400(根据实施例1制备的)上的降解动力学。
对于so降解(图17),观察到au-pd(1:1)/nm-400对so的降解速率高于nm300、au/nm-400和pd/nm-400的降解速率。此外,对于nm300、au/nm-400、pd/nm-400和au-pd(1:1)/nm-400,so降解遵循准一级反应动力学,其反应速率常数分别为0.20、0.24、0.23和0.46min-1。如图18和图19所示,还观察到了mb和bg的相似降解动力学,它们也遵循准一级反应动力学。表4中总结了用于降解每种染料的每种催化剂的反应速率常数。
表4.nm300、au/nm-400、pd/nm-400和au-pd(1:1)/nm-400分别在降解so、mb和bg中的反应速率常数。
实施例11.所提出的au-pd(1:1)/nm-400上染料降解的催化机理
推断出活性氧类(ros)在通过本发明的催化剂的染料的催化降解中起作用。为了进一步了解ros在au-pd(1:1)/nm-400上染料降解机理中的作用,1,4-苯醌(bq)、nan3和二甲基亚砜(dmso)分别用作为超氧自由基(·o2-)、单线态氧(1o2)和羟基自由基(·oh)的淬灭剂(p.wang,t.-t.lim,waterres.,2012,46,1825-1837)。
如图20a所示,在ros淬灭剂的存在下,so降解受到抑制,其显著性顺序依次为bq>nan3>dmso。对于mb和bg降解(分别为图20b和图20c)观察到类似的现象,其中·o2-和1o2两者显著促进了染料降解而发现·oh则没有那么显著。因此,通过超氧自由基机理和单个氧机理推断·o2-和1o2是染料的cwao期间的主要活性氧化类。
如上所述,xps分析证实,au-pd沉积在催化剂载体上后,催化剂表面吸附的氧类增加,并且对于pd0的电子略有损失(图5)。此外,在载体表面上还应该产生痕量的o-空位,其可能分布在au-pd和moo3之间的界面处。
基于上述淬火实验和xps分析,提出的通过使用au-pd(1:1)/nm-400降解染料的催化机理如图20d所示。通常,au-pd合金颗粒表面上的电子可以被部分转移到位于au-pd和moo3之间的界面处的moo3中的相邻o-空位点(h.zhang,等人,nat.mater.,2011,11,49),然后通过o空位点上吸附的o2俘获所述电子以形成·o2-和1o2(h.masatake,等人,chem.lett.,1987,16(1987)405-408;m.r.hoffmann,等人,chem.rev.,1995,95,69-96)。所产生的·o2-和1o2具有用于有机化合物的降解的潜力。
实施例12.将双金属复合催化剂引入废水处理系统的可能方式
可以通过各种方式将双金属复合催化剂体系引入水处理工艺中。一种方式是使催化剂自由地悬浮在废物流中,如图21a所示。废物流入物20将被添加有自由悬浮的双金属复合催化剂40。与空气或o2(60)的气泡一起,将废水净化以产生产物水80,而将分散的催化剂40从产物水80中分离出来并回收以重复使用。
在其他形式中,如图21b所示,双金属复合催化剂40在一定范围的多孔材料100(例如,网格、纤维、织物和膜)上可以是固定的或不动的状态。在空气或o2(60)的气泡的情况下,废水20可以被净化以产生净化水80。通过去除包含催化剂40的多孔载体100,该结构可以促进从净化水中容易地分离催化剂。
- 还没有人留言评论。精彩留言会获得点赞!