用于可调基底生长的多壁碳纳米管的催化剂和工艺的制作方法
背景
目前,氢气似乎是最有前途和环境友好的能源,因为它可以以较少的污染和高的效率被转化成电力和其他能源形式。在多种氢气生产方法中,天然气的蒸汽重整是最受欢迎和最经济的技术,其占世界氢气消耗量的50%(j.holladay等人,catal.today,2009,139,244–260)。但是这些工艺是高度吸热的(68.7kjmol-1h2)并且产生大量的cox。结果,蒸汽重整伴随着水煤气变换反应、分离以及纯化步骤,从而增加了工艺的成本。最近,页岩气已经成为美国非常重要的天然气来源。在2005年,其仅占美国天然气产量的3%,到2012年上升至35%,并且预测到2035年将增长至近50%(r.w.howarth,energysci.eng.,2014,2,47–60)。对无cox的氢气的需求的增加和页岩气资源的丰富为开发新的化学工艺提供了机会,该化学工艺将其主要成分甲烷转化为更有价值的燃料和化学品。
甲烷的直接转化是有吸引力的替代工艺,因为它与蒸汽重整相比是吸热较少的工艺(37.4kjmol-1h2;参见j.n.armor,appl.catal.agen.,1999,176,159–176;q.weizhong等人,appl.catal.agen.,2004,260,223–228)。该工艺产生无cox的氢气,其在低温燃料电池中具有很大的应用,并且还产生有价值的多个碳纳米管(碳纳米管)或纳米纤维。甲烷,一种直链烃,可热分解为碳原子,其最终通过以下反应形成直的和中空的碳纳米管:
尽管已经进行了广泛的研究以确定用于通过甲烷分解来生产碳纳米管的工艺,但是目前这些方法中没有一种对于工业规模生产是可行的。在所使用的金属催化剂中,由于基于ni的催化剂的高催化活性和生产碳纳米管的能力,大多数研究人员已经集中在基于ni的催化剂上(t.zhang和m.d.amiridis,appl.catal.agen.,1998,167,161–172;a.m.amin等人,int.j.hydrogenenergy,2011,36,2904–2935)。尽管ni催化剂显示出优异的性能,但它们在高于600℃的温度立即失活。(s.t.hussain等人,j.nat.gaschem.,2008,17,374–382;y.wang等人,int.j.hydrogenenergy,2014,39,778–787)。为了改善耐久性并且减少催化剂在反应温度的失活,不同的金属和金属氧化物已经被引入到基于ni的催化剂中(g.wang等人,energy&fuels,2013,27,4448–4456;y.li等人,chem.commun.,1999,1141–1142;j.chen等人,appl.catal.agen.,2004,269,179–186;i.suelves等人,catal.today,2006,116,271–280;j.ashok等人,energy&fuels,2009,23,5–13;a.monzel等人,catal.today,2006,116,264–270)。还已经研究了基于铁的催化剂,但是其显示出比ni催化剂更短的寿命和更低的活性(k.g.ermakovaandermakovd.y.,appl.catal.agen.,2000,201,61–70)。当使用基于fe的催化剂时,还需要较高的温度范围用于高效操作。与含ni和fe的催化剂相比,co催化剂已经受到较少的关注,但仍然存在几个示出他们对甲烷分解的活性的文献(a.e.awadallah等人,chem.eng.commun.,2015,202,163–174;l.b.avdeeva等人,appl.catal.agen.,2002,228,53–6;a.h.fakeeha等人,pet.sci.technol.,2016,34,1617–1623)。基于先前的文献,可以总结出铁族金属的催化活性依次为ni>co>fe(l.b.avdeeva等人,appl.catal.agen.,2002,228,53–6)。不幸的是,由于通过所形成的碳覆盖了活性位点,催化剂的活性在反应过程期间逐渐丧失。
目前可用的催化甲烷分解的方法通过“尖端生长”形成碳纳米管。然而,尖端生长的碳纳米管的显著缺点是,在使用酸或碱处理从催化剂中收获碳纳米管的过程期间,金属纳米颗粒被溶解并且催化剂被牺牲。此外,尖端生长的碳纳米管导致附着至尖端的催化剂纳米颗粒,在碳纳米管中产生杂质。因此,在完全回收催化剂的情况下提取碳纳米管是最推荐的。为了克服该问题,催化剂和反应过程以产生“基底生长的(basegrown)”碳纳米管的这样的方式被设计。据信,基底生长的碳纳米管可以容易地被收获并且催化剂可以被再生,而不会在提取过程期间产生消耗。
尽管在针对伴随生产碳纳米管的无cox的氢气生产的研究中有进展,但是仍然缺乏实现这些生产的组合物和方法,特别是以允许从所使用的催化剂中容易地分离所生产的碳纳米管的方式来实现这些生产的组合物和方法。这些需求和其他需求通过本公开内容来满足。
概述
根据本公开内容的目的,如本文所体现的和广泛描述的,本公开内容在一个方面涉及用于利用所公开的催化剂从低级烃同时生产碳纳米管和氢气的方法和组合物,所述低级烃包括甲烷、乙烷、丙烷、丁烷或其组合。在多个方面,本公开内容涉及用于伴随生产碳纳米管的无cox的氢气生产的方法。还公开了用于在所公开的催化剂组合物上的选择性的基底生长的碳纳米管的方法和组合物。在另外的方面,本公开内容涉及包含在选自二氧化硅、氧化铝、沸石、二氧化钛及其组合的载体材料上的3d过渡金属(例如,ni、fe、co、mn、cr、mo及其组合)的单金属催化剂、双金属催化剂和三金属催化剂。还公开了用于制备所公开的催化剂的方法。
公开了包含两种3d过渡金属和载体材料的组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自3d过渡金属,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石或其混合物。
公开了包含两种3d过渡金属和载体材料的组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自ni、fe、co、mn、cr、mo及其组合,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石或其混合物。
还公开了包含两种3d过渡金属和载体材料的组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自3d过渡金属,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石或其混合物。
还公开了包含3d过渡金属和载体材料的组合物,其中3d过渡金属选自ni、fe、co、mn、cr、mo及其组合;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在。
还公开了包含3d过渡金属和载体材料的组合物,其中3d过渡金属选自ni、fe和co;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在。
还公开了制备所公开的组合物的方法,包括初湿含浸技术(incipientwetnesstechnique)。
还公开了制备所公开的组合物的方法,包括溶胶-凝胶技术。
还公开了制备所公开的组合物的方法,该方法包括以下步骤:使载体材料与包含第一3d过渡金属盐和第二3d过渡金属的水溶液接触,以形成包含载体材料以及包含第一3d过渡金属盐和第二3d过渡金属金属的水溶液的混合物;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;并且其中第一3d金属盐和第二3d金属盐不相同;干燥混合物以形成干燥的混合物;煅烧所述干燥的混合物以形成煅烧的混合物;以及还原所述煅烧的混合物,从而提供包含两种3d过渡金属和载体材料的组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自3d过渡金属,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石或其混合物。
还公开了制备所公开的组合物的方法,该方法包括以下步骤:使载体材料与包含第一3d过渡金属盐和第二3d过渡金属的水溶液接触,以形成包含载体材料以及包含第一3d过渡金属盐和第二3d过渡金属金属的水溶液的混合物;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;并且其中第一3d金属盐和第二3d金属盐不相同;干燥混合物以形成干燥的混合物;煅烧所述干燥的混合物以形成煅烧的混合物;以及还原所述煅烧的混合物,从而提供包含两种3d过渡金属和载体材料的组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自ni、fe、co、mn、cr、mo及其组合,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石或其混合物。
还公开了制备包含3d过渡金属和载体材料的组合物的方法,其中3d过渡金属选自ni、fe、co、mn、cr、mo及其组合;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在;该方法包括以下步骤:使载体材料与包含3d过渡金属盐的水溶液接触,所述3d过渡金属盐选自镍盐、铁盐、钴盐、锰盐、铬盐、钼盐或其组合;形成包含载体材料和包含3d过渡金属盐的水溶液的混合物;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中3d过渡金属盐以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在。
还公开了制备包含3d过渡金属和载体材料的组合物的方法,其中3d过渡金属选自ni、fe和co;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在;该方法包括以下步骤:使载体材料与包含3d过渡金属盐的水溶液接触,所述3d过渡金属盐选自镍盐、铁盐和钴盐;形成包含载体材料和包含3d过渡金属盐的水溶液的混合物;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中3d过渡金属盐以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在。
还公开了分解低级烃的方法,该方法包括以下步骤:以约1℃min-1至约20℃min-1的加热速率将催化剂床加热到约500℃至约1000℃的温度;其中催化剂床包括所公开的组合物;其中所公开的组合物包含两种3d过渡金属和载体材料,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自3d过渡金属,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中催化剂床被定位在固定流动反应器的反应器床内;其中固定流动反应器包括用于气体流的入口端(inletend)和出口端(outletend);基于约0.1克催化剂负载量,以约25mlmin-1至约200mlmin-1的惰性气体流量通过入口端提供第一惰性气体的流(除非另有指示,否则在本申请中给出的所有气体流量均是基于0.1g催化剂负载量);其中第一惰性气体的流与催化剂床接触;终止第一惰性气体的流通过入口端;以等于约5,000h-1至约60,000h-1的空速(spacevelocity)的反应物气体流量和从约0分钟至约240分钟的运行时间(time-on-stream)(tos)提供通到入口端的反应物气体的流;其中反应物气体的流与催化剂床接触;并且其中反应物气体包括低级烃和第二惰性气体;在出口处收集流出气体;其中流出气体包括氢气。
还公开了分解低级烃的方法,该方法包括以下步骤:以约1℃min-1至约20℃min-1的加热速率将催化剂床加热到约500℃至约1000℃的温度;其中催化剂床包括所公开的组合物;其中所公开的组合物包含两种3d过渡金属和载体材料,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自ni、fe、co、mn、cr、mo及其组合,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中催化剂床被定位在固定流动反应器的反应器床内;其中固定流动反应器包括用于气体流的入口端和出口端;基于约0.1克催化剂负载量,以约25mlmin-1至约200mlmin-1的惰性气体流量通过入口端提供第一惰性气体的流(除非另有指示,否则在本申请中给出的所有气体流量均是基于0.1g催化剂负载量);其中第一惰性气体的流与催化剂床接触;终止第一惰性气体的流通过入口端;以等于约5,000h-1至约60,000h-1的空速的反应物气体流量和从约0分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流;其中反应物气体的流与催化剂床接触;并且其中反应物气体包括低级烃和第二惰性气体;在出口处收集流出气体;其中流出气体包括氢气。
还公开了分解低级烃的方法,该方法包括以下步骤:以约1℃min-1至约20℃min-1的加热速率将催化剂床加热到约500℃至约1000℃的温度;其中催化剂床包括所公开的组合物;其中所公开的组合物包含3d过渡金属和载体材料,其中3d过渡金属选自ni、fe、co、mn、cr、mo及其组合;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中催化剂床被定位在固定流动反应器的反应器床内;其中固定流动反应器包括用于气体流的入口端和出口端;以约25mlmin-1至约200mlmin-1的惰性气体流量通过入口端提供第一惰性气体的流;其中第一惰性气体的流与催化剂床接触;终止第一惰性气体的流通过入口端;以等于约5,000h-1至约60,000h-1的空速的反应物气体流量和从约0分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流;其中反应物气体的流与催化剂床接触;并且其中反应物气体包括低级烃和第二惰性气体;在出口处收集流出气体;其中流出气体包括氢气。
还公开了分解低级烃的方法,该方法包括以下步骤:以约1℃min-1至约20℃min-1的加热速率将催化剂床加热到约500℃至约1000℃的温度;其中催化剂床包括所公开的组合物;其中所公开的组合物包含3d过渡金属和载体材料,其中3d过渡金属选自ni、fe和co;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中催化剂床被定位在固定流动反应器的反应器床内;其中固定流动反应器包括用于气体流的入口端和出口端;以约25mlmin-1至约200mlmin-1的惰性气体流量通过入口端提供第一惰性气体的流;其中第一惰性气体的流与催化剂床接触;终止第一惰性气体的流通过入口端;以约25mlmin-1至约200mlmin-1的反应物气体流量和约20,000h-1至约60,000h-1的空速以及从约0分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流;其中反应物气体的流与催化剂床接触;并且其中反应物气体包括低级烃和第二惰性气体;在出口处收集流出气体;其中流出气体包括氢气。
虽然本公开内容的方面可以在特定的法定类别(诸如系统法定类别)中被描述和被要求保护,但是这仅仅是为了方便,并且本领域技术人员将理解,本公开内容的每个方面可以在任何法定类别中被描述和被要求保护。除非另有明确陈述,否则决不意图本文阐述的任何方法或方面被解释为需要以特定的顺序执行其步骤。因此,在方法权利要求未在权利要求或描述中具体陈述步骤将被限制为特定顺序的情况下,无论如何都决不意图推断顺序。这适用于解释的任何可能的非明示基础,包括与步骤的安排或操作流程相关的逻辑问题,源自语法组织或标点符号的简单含义,或说明书中描述的方面的数量或类型。
附图简述
被并入到本说明书中并构成本说明书的一部分的附图例证了若干方面,并与描述一起用来解释本公开内容的原理。
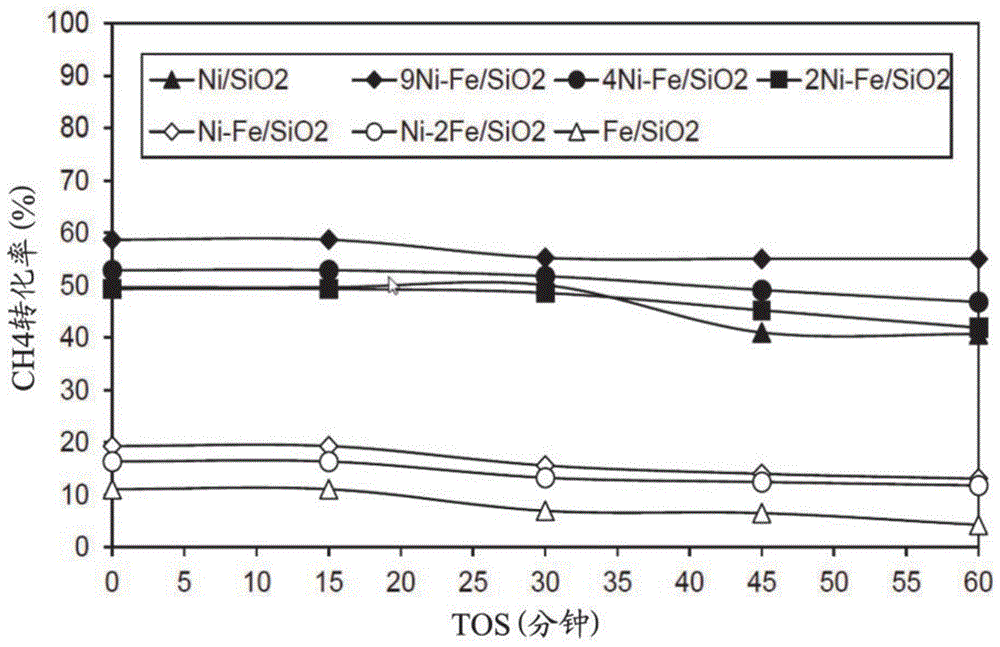
图1示出了在t=650℃、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在具有多种摩尔比的代表性公开的ni-fe/sio2催化剂上的甲烷分解的代表性数据。
图2示出了在t=650℃、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在具有多种摩尔比的代表性公开的ni-co/sio2催化剂上的甲烷分解的代表性数据。
图3示出了在t=650℃、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在具有多种摩尔比的代表性公开的fe-co/sio2催化剂上的甲烷分解的代表性数据。
图4a-图4c示出了在代表性公开的单金属催化剂和双金属催化剂上的碳纳米管的x射线衍射图的代表性数据。具体而言,图4a示出了在代表性公开的ni-fe催化剂上的碳纳米管的x射线衍射图的代表性数据。图4b示出了在代表性公开的ni-co催化剂上的碳纳米管的x射线衍射图的代表性数据。图4c示出了在代表性公开的fe-co催化剂上的碳纳米管的x射线衍射图的代表性数据。
图5示出了沉积在代表性公开的9ni-1fe/sio2催化剂、9ni-1co/sio2催化剂和1fe-2co/sio2催化剂上的碳的代表性热稳定性数据。
图6示出了根据图中的标记的在代表性公开的单金属和双金属ni-fe催化剂上的碳纳米管的代表性拉曼光谱数据。
图7示出了根据图中的标记的在代表性公开的单金属和双金属ni-co催化剂上的碳纳米管的代表性拉曼光谱数据。
图8示出了根据图中的标记的在代表性公开的单金属和双金属fe-co催化剂上的碳纳米管的代表性拉曼光谱数据。
图9a-图9f示出了在代表性公开的催化剂上的碳丝的生长的代表性图像。具体而言,图9a和图9b示出了在不同放大倍数(参见图中的比例尺)在代表性公开的9ni-1fe/sio2催化剂上的碳丝的生长的代表性图像;图9c和图9d示出了在不同放大倍数(参见图中的比例尺)在代表性公开的9ni-1co/sio2催化剂上的碳丝的生长的代表性图像;以及图9e和图9f示出了在代表性公开的1fe-2co/sio2催化剂上的碳丝的生长的代表性图像。所有图像都是在t=650℃、tos=60分钟、ghsv=42000h-1的条件下如在实例中所描述进行的甲烷分解之后获得的。
图10示出了在指示的温度和tos=60分钟/ghsv=42000h-1下,在fe/sio2催化剂上的碳丝的基底生长的代表性数据。
图11a和图11b示出了在t=700℃、tos=60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在代表性公开的fe/sio2催化剂上的基底生长的碳纳米管的代表性透射电子显微照片。
图12示出了甲烷转化为氢气的代表性数据,该数据是从在t=650℃、tos=60分钟、ghsv=42000h-1的条件下如在实例中所描述进行的对代表性公开的fe/sio2催化剂的催化剂再生研究获得的。
图13a和图13b示出了在t=700℃、tos=60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在fe/sio2催化剂的第二个循环中的基底生长的碳纳米管的代表性透射电子显微照片。
图14示出了从在fe/sio2催化剂上制备的基底生长的碳纳米管获得的代表性拉曼光谱数据。线(a)示出了从在第一反应循环期间制备的碳纳米管获得的数据;并且线(b)示出了从在再生后的第二反应循环期间制备的碳纳米管获得的数据。反应条件为t=700℃、tos=60分钟、ghsv=42000h-1。
图15a和图15b示出了如所指示的新鲜的代表性公开的单金属和双金属ni-fe催化剂的代表性x射线衍射数据。
图16示出了如所指示的用过的代表性公开的单金属和双金属ni-fe催化剂的代表性x射线衍射数据。
图17a和图17b示出了如所指示的新鲜的代表性公开的单金属和双金属ni-co催化剂的代表性x射线衍射数据。
图18示出了如所指示的用过的代表性公开的单金属和双金属ni-co催化剂的代表性x射线衍射数据。
图19a和图19b示出了如所指示的新鲜的代表性公开的单金属和双金属fe-co催化剂的代表性x射线衍射数据。
图20示出了如所指示的用过的代表性公开的单金属和双金属fe-co催化剂的代表性x射线衍射数据。
图21示出了从如所指示的新鲜的ni-fe催化剂的程序升温还原分析(temperature-programmedreductionanalysis)获得的代表性数据。
图22示出了从如所指示的新鲜的ni-co催化剂的程序升温还原分析获得的代表性数据。
图23示出了从如所指示的新鲜的fe-co催化剂的程序升温还原分析获得的代表性数据。
图24示出了在t=650℃、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在具有多种摩尔比(如所指示的)的代表性公开的ni-fe/sio2催化剂上的h2收率的代表性数据。
图25示出了在t=650℃、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在具有多种摩尔比(如所指示的)的代表性公开的ni-fe/sio2催化剂上形成的碳的量的代表性数据。
图26示出了在t=650℃、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在具有多种摩尔比(如所指示的)的代表性公开的ni-co/sio2催化剂上的h2收率的代表性数据。
图27示出了在t=650℃、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在具有多种摩尔比(如所指示的)的代表性公开的ni-co/sio2催化剂上形成的碳的量的代表性数据。
图28示出了在t=650℃、p=1巴、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在具有多种摩尔比(如所指示的)的代表性公开的fe-co/sio2催化剂上的h2收率的代表性数据。
图29示出了在t=650℃、p=1巴、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在具有多种摩尔比(如所指示的)的代表性公开的fe-co/sio2催化剂上形成的碳的量的代表性数据。
图30示出了在t=650℃、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在还原形式和氧化形式的代表性公开的9ni-1fe/sio2催化剂上的甲烷分解的代表性数据。
图31示出了在t=650℃、p=1巴、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在代表性公开的9ni-1co/sio2催化剂上的尖端生长的碳纳米管和基底生长的碳纳米管的代表性图像。比例尺在图中指示。
图32示出了在t=650℃、p=1巴、tos=60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在代表性公开的1fe-2co/sio2催化剂上的尖端生长的碳纳米管和基底生长的碳纳米管的代表性图像。比例尺在图中指示。
图33a和图33b示出了在t=650℃、p=1巴、tos=0-60分钟、ghsv=42000h-1的条件下,如在实例中所描述进行的在代表性公开的fe/al2o3催化剂(60wt%fe/40wt%al2o3)上的碳纳米管的代表性图像。比例尺在图中指示。
图34示出了所指示的代表性的催化剂/载体样品的甲烷转化效率的代表性数据。“700℃60fe/al2o3”指示催化剂/载体体系为60wt%fe/40wt%al2o3,其中甲烷的分解如在实例中所描述的在t=700℃、p=1巴、tos=0-60分钟、ghsv=42000h-1的条件下进行。“650℃60fe/al2o3”指示催化剂/载体体系为60wt%fe/40wt%al2o3,其中甲烷的分解如在实例中所描述的在t=650℃、p=1巴、tos=0-60分钟、ghsv=42000h-1的条件下进行。“700℃60fe/sio2”指示催化剂/载体体系为60wt%fe/40wt%sio2,其中甲烷的分解如在实例中所描述的在t=700℃、p=1巴、tos=0-60分钟、ghsv=42000h-1的条件下进行。“700℃60fe/沸石”指示催化剂/载体体系为60wt%fe/40wt%沸石,其中甲烷的分解如在实例中所描述的在t=700℃、p=1巴、tos=0-60分钟、ghsv=42000h-1的条件下进行。
图35示出了在t=700℃、tos=0-60分钟,用于在多种载体上的所公开的fe催化剂上的甲烷分解的代表性数据。
图36示出了在t=700℃、tos=0-60分钟,用于在具有多种fe负载量的所公开的fe/al2o3催化剂上的甲烷分解的代表性数据。
图37a-图37d示出了与用于分离和纯化所公开的碳纳米管的酸回流方法的效果有关的代表性数据。图37a示出了用过的所公开的9ni:1fe/sio2和fe/sio2催化剂以及在酸回流之后纯化的碳纳米管的代表性的xrd图数据。图37b示出了用过的所公开的9ni:1fe/sio2和fe/sio2催化剂以及在酸回流之后纯化的碳纳米管的代表性的拉曼光谱数据。图37c示出了在酸回流之前所公开的碳纳米管的代表性透射电子显微图像。图37d示出了在酸回流之后所公开的碳纳米管的代表性透射电子显微图像。
图38a-图38b示出了所公开的介孔气凝胶催化剂的提出的形成机理的示意图。图38a示出了所公开的介孔气凝胶催化剂ni/al2o3的提出的形成机理。图38b示出了所公开的介孔气凝胶催化剂co/al2o3的提出的形成机理。
图39a-图39d示出了在所公开的金属氧化物气凝胶催化剂上的甲烷分解的代表性数据。对于所示出的数据,t=650℃、p=0.1mpa并且ghsv=42000h-1。图39a示出了在具有如图中所描述的wt%ni的量的公开的ni/al2o3气凝胶催化剂上的甲烷转化率。图39b示出了在具有如图中所描述的wt%ni的量的公开的ni/al2o3气凝胶催化剂上的h2收率。图39c示出了在具有如图中所描述的wt%ni的量的公开的co/al2o3气凝胶催化剂上的甲烷转化率。图39d示出了在具有如图中所描述的wt%ni的量的公开的co/al2o3气凝胶催化剂上的h2收率。
图40a-图40d示出了在所公开的金属氧化物气凝胶催化剂上的甲烷分解的代表性数据,与通过初湿含浸法制备的金属氧化物催化剂进行比较。对于所示出的数据,t=650℃并且p=0.1mpa1。使用图中公开的气凝胶催化剂获得的数据对应于与50wt%ni/al2o3、60wt%ni/al2o3和70wt%ni/al2o3相关联的线。图中所指示的其他单金属金属氧化物催化剂或双金属金属氧化物催化剂通过初湿含浸法制备。图40a、图40b、图40c、图40d、图40e和图40f中的每一个示出了与通过初湿含浸法制备的所指示的金属氧化物催化剂相比的在所公开的金属氧化物气凝胶催化剂(50wt%ni/al2o3、60wt%ni/al2o3和70wt%ni/al2o3)上的甲烷转化率数据。
图41a-41b示出了在如所指示的新鲜的和用过的所公开的气凝胶催化剂上获得的代表性数据x射线衍射(xrd)分析数据。图41a示出了在新鲜的和用过的所公开的气凝胶催化剂ni/al2o3上获得的xrd数据。图41a示出了在新鲜的和用过的所公开的气凝胶催化剂co/al2o3上获得的xrd数据。
图42a-图42d示出了在使用之后的所公开的气凝胶催化剂的代表性数据。图42a示出了从所公开的使用的ni/al2o3气凝胶催化剂获得的代表性拉曼光谱数据。图42b示出了从所公开的使用的co/al2o3气凝胶催化剂获得的代表性拉曼光谱数据。图42c示出了从所公开的使用的ni/al2o3气凝胶催化剂获得的代表性x射线光电子能谱(xps)数据。图42d示出了从所公开的使用的co/al2o3气凝胶催化剂获得的代表性x射线光电子能谱(xps)数据。
图43a-图43d示出了与使用之后的公开的气凝胶催化剂60wt%ni/al2o3和co/al2o3气凝胶催化剂的表征有关的代表性数据。图43a示出了所指示的公开的气凝胶催化剂的n2吸收-解吸等温线(absorption-desorptionisotherm)的brunauer-emmett-teller(bet)分析。图43b示出了所指示的公开的气凝胶催化剂的孔径分布数据。图43c和图43d各自示出了公开的气凝胶催化剂60wt%ni/al2o3的代表性扫描电子显微镜(sem)图像。图43c和图43d中的图像处于不同的放大倍数,其中比例尺如在每个图像中所示。
图44a-图44f示出了公开的气凝胶催化剂的代表性透射电子显微镜(tem)图像。图44a、图44b和图44c中的每一个示出了所公开的60wt%ni/al2o3气凝胶催化剂的代表性tem图像,而图44d、图44e和图44f中的每一个示出了所公开的70wt%ni/al2o3气凝胶催化剂的代表性tem图像。在多个图中,比例尺在每个图的左下方示出并且碳纳米管尖端结构由带虚线的圆圈突出显示。
图45a-图45f示出了所公开的气凝胶催化剂的代表性透射电子显微镜(tem)图像。图45a、图45b和图45c中的每一个示出了所公开的60wt%co/al2o3气凝胶催化剂的代表性tem图像,而图45d、图45e和图45f中的每一个示出了所公开的70wt%co/al2o3气凝胶催化剂的代表性tem图像。在多个图中,比例尺在每个图的左下方示出并且碳纳米管尖端结构由带虚线的圆圈突出显示。
图46a-图46b示出了在多个再生循环之后再生的公开的气凝胶催化剂的代表性数据。图46a示出了在650℃、tos=60分钟和ghsv=42000h-1,在公开的60wt%ni/al2o3气凝胶催化剂上的催化剂再生研究中的甲烷转化率结果。在催化剂再生研究中,在每个循环之后,通过在650℃用10%的o2处理催化剂持续45分钟来再生催化剂。图46b示出了在650℃、tos=60分钟和ghsv=42000h-1,在公开的60wt%ni/al2o3气凝胶催化剂上的催化剂再生研究中的h2收率结果。在催化剂再生研究中,在每个循环之后,通过在650℃用10%的o2处理催化剂持续45分钟来再生催化剂。
图47a-图47d示出了所公开的气凝胶催化剂的代表性数据代表性透射电子显微镜(tem)图像。图47a、图47b、图47c和图47d中的每一个示出了在图46a-图46b中描述的研究的第二循环之后的60wt%ni/al2o3气凝胶催化剂的代表性tem图像。在多个图中,比例尺在每个图的左下方示出并且碳纳米管尖端结构由带虚线的圆圈突出显示。
图48a-图48d示出了所公开的气凝胶催化剂的代表性数据代表性透射电子显微镜(tem)图像。图48a、图48b、图48c和图48d中的每一个示出了来自在图46a-图46b中描述的研究的第五循环之后的60wt%ni/al2o3气凝胶催化剂的代表性tem图像。在多个图中,比例尺在每个图的左下方示出并且碳纳米管尖端结构由带虚线的圆圈突出显示。
本公开内容的另外的优点将在以下描述中部分地阐述,并且从描述中将部分地是明显的或者可以通过实践本公开内容而获知。本公开内容的优点将通过在所附权利要求中具体指出的要素和组合被实现和获得。应理解,前述一般性描述和以下详细描述两者都仅是示例性的和解释性的,并且不限制如所要求保护的本公开内容。
详述
下文将参考附图更全面地描述本文的公开内容,在附图中示出了一些但不是所有的可能的实施方案。事实上,本公开内容可以以许多不同的形式来体现并且不应当被解释为限于本文阐述的实施方案;相反,提供这些实施方案使得本公开内容将满足适用的法律要求。相同的数字在全文中指代相同的要素。
受益于在前面的描述和相关的附图中呈现的教导,所公开的组合物和方法所属领域中的技术人员将会想到本文公开的许多修改和其他实施方案。因此,应当理解,本公开内容不限于所公开的具体实施方案,并且修改和其他实施方案被意图包括在所附权利要求的范围内。虽然本文使用了特定术语,但它们仅用于一般性和描述性的意义,并且不用于限制的目的。
还应当理解,本文使用的术语仅是出于描述特定方面的目的,并且不意图是限制性的。如在说明书中和在权利要求书中所使用的,术语“包括(comprising)”可以包括“由……组成”的方面。除非另有定义,否则本文使用的所有技术术语和科学术语都具有与所公开的组合物和方法所属领域的普通技术人员通常理解的相同的含义。在本说明书中和在所附的权利要求书中,将参考将在本文中定义的多个术语。
如对于本领域技术人员在阅读本公开内容时将明显的是,本文所描述和示出的单独的实施方案中的每个具有离散的组分(component)和特征,这些组分和特征可以容易地与任何其他若干实施方案的特征分离或组合,而不偏离本公开内容的范围或精神。任何陈述的方法可以以所陈述的事件的顺序或以逻辑上可能的任何其他顺序进行。
本文提及的所有出版物都通过引用并入本文,以公开和描述与引用出版物相关的方法和/或材料。本文讨论的出版物仅为了其在本申请的申请日之前的公开内容而提供。本文的任何内容都不应被解释为承认本发明无权凭借在先发明而先于这样的出版物。此外,本文提供的出版物的日期可以不同于可能需要单独确认的实际公布日期。
在描述多种实施方案之前,除非另有指示,否则提供并且应当使用以下定义。
除非另外定义,否则本文使用的所有技术术语和科学术语具有与本公开内容所属领域的普通技术人员通常理解的相同的含义。还应该理解,术语,诸如那些在常用词典中定义的术语,应该被解释为具有与它们在说明书和相关领域的上下文中的含义一致的含义,并且不应该以理想化的或过于正式的意义来解释,除非本文明确定义。
如本文中所使用的,化合物(包括有机化合物)的命名可以使用通用名称、iupac、iubmb或cas命名推荐来给出。当存在一个或更多个立体化学特征时,用于立体化学的cahn-ingold-prelog规则可以用于指定立体化学优先级、e/z规格及其他。如果给定名称,通过使用命名约定来系统地简化化合物结构,或者通过商业上可获得的软件诸如chemdrawtm(cambridgesoftcorporation,u.s.a.),本领域技术人员可以很容易地确定化合物的结构。
如本文中所使用的,“包括”将被解释为指定如所提及的陈述的特征、整数、步骤或组分的存在,但不排除一个或更多个特征、整数、步骤或组分或其组的存在或添加。此外,术语“包括”旨在包括由术语“基本上由……组成”和“由……组成”所涵盖的实例。类似地,术语“基本上由……组成”旨在包括由术语“由……组成”所涵盖的实例。
如在本说明书和所附权利要求中所使用的,除非上下文中另外清楚地指示,否则单数形式“一(a)”、“一(an)”和“该(the)”包括复数指示物。因此,例如,提及“一种碳纳米管”、“一种催化剂”或“一种基底”,包括但不限于两种或更多种这样的碳纳米管、催化剂或基底及类似物。
提及“一种”化合物是指该化合物的一个或更多个分子,而不是限于该化合物的单个分子。此外,一个或更多个分子可以相同或可以不同,只要它们属于化合物的范畴。因此,例如,“一种”fe-co催化剂被解释为包括一种或更多种催化剂组合物,其中催化剂组合物可以相同或可以不同(例如,不同比率的fe-co)。
应当注意,比率、浓度、量和其他数值数据在本文中可以以范围格式表述。在所陈述的范围包括限值中的一个或两个的情况下,在本公开内容中还包括排除那些所包括的限值中的任一个或两个的范围,例如措辞“x至y”包括从‘x’到‘y’的范围以及大于‘x’且小于‘y’的范围。该范围还可以被表示为上限,例如‘约x、y、z或更小’,并且应该被解释为包括‘约x’、‘约y’和‘约z’的特定范围以及‘小于x’、‘小于y’和‘小于z’的范围。同样,措辞‘约x、y、z或更大’应当被解释为包括‘约x’、‘约y’和‘约z’的特定范围以及‘大于x’、‘大于y’和‘大于z’的范围。此外,措辞“约‘x’至‘y’(其中‘x’和‘y’是数值)包括“约‘x’至约‘y’”。应当理解,这样的范围格式是为了方便和简洁起见而使用的,并且因此应当以灵活的方式被解释为不仅包括作为范围的限值明确陈述的数值,而且还包括在该范围内涵盖的所有单独的数值或子范围,如同每个数值和子范围被明确地陈述。为了说明,“约0.1%至约5%的数值范围应当被解释为不仅包括约0.1%至约5%的明确陈述的值,而且还包括在所指示的范围内的单独的值(例如,1%、2%、3%和4%)和子范围(例如,0.5%、1.1%、2.2%、3.3%和4.4%)。
如本文中所使用的,术语“约”、“近似”、“处于或约”和“基本上”意指所讨论的量或值可以是精确值或提供如权利要求中所陈述的或本文中所教导的等效结果或效果的值。也就是,应当理解,量、大小、配方、参数和其他数量和特性不是精确的并且不需要是精确的,但是根据需要可以是近似的和/或更大的或更小的,反映了公差、转换因子、四舍五入、测量误差及类似的,以及本领域技术人员已知的其他因素,使得获得等效结果或效果。在一些情况下,提供等效结果或效果的值无法被合理地确定。在这样的情况下,如本文中所使用的,通常应当理解,“约”和“处于或约”意指指示的标称值±10%变化,除非另有指示或推断。通常,量、大小、配方、参数或其他数量或特性是“约”、“近似”或“处于或约”,无论是否明确地陈述是这样的。应当理解,在定量值之前使用“约”、“近似”或“处于或约”的情况下,参数还包括具体的定量值本身,除非另有具体陈述。
金属负载的催化剂
在多个方面,本公开内容涉及金属负载的催化剂组合物,该金属负载的催化剂组合物具有用于利用所公开的催化剂从甲烷同时生产碳纳米管和氢气的效用。
在多个方面,本公开内容涉及包含在选自二氧化硅、氧化铝、沸石或其混合物的载体材料上的3d过渡金属(例如,ni、fe、co、mn、cr、mo及其组合)的三金属催化剂。
在多个方面,本公开内容涉及包含在选自二氧化硅、氧化铝、沸石或其混合物的载体材料上的3d过渡金属(例如,ni、fe、co、mn、cr、mo及其组合)的双金属催化剂。
在多个方面,本公开内容涉及包含在选自二氧化硅、氧化铝、沸石或其混合物的载体材料上的3d过渡金属(例如,ni、fe、co、mn、cr和mo)的单金属催化剂。
在多个方面,本文公开了包含两种3d过渡金属和载体材料的催化剂组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自3d过渡金属,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石或其混合物。
在多个方面,本文公开了包含两种3d过渡金属和载体材料的催化剂组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自ni、fe、co、mn、cr、mo及其组合,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石或其混合物。
在多个方面,本文公开了包含两种3d过渡金属和载体材料的催化剂组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自3d过渡金属,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石或其混合物。
在多个方面,本文公开了包含3d过渡金属和载体材料的催化剂组合物,其中3d过渡金属选自ni、fe、co、mn、cr、mo及其组合;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在。
在多个方面,本文公开了包含3d过渡金属和载体材料的催化剂组合物,其中3d过渡金属选自ni、fe和co;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在。
在多个方面,所公开的催化剂使用如本领域技术人员已知的初湿含浸技术来制备。在其他方面,所公开的催化剂是使用溶胶-凝胶技术制备的气凝胶催化剂,例如,如本文所描述的或如本领域技术人员通常已知的。
如本文中所使用的,术语“初湿含浸”通常是指自由流动的多孔粉末变得润湿到这样的程度,即液体的另外的少量添加导致其流动能力的显著降低(例如,参见w.b.innes,analyticalchemistry,第28卷,第3期,1956年3月,第332页,其通过引用并入本文)。
如本文中所使用的,“气凝胶催化剂”是包含至少一种或更多种3d过渡金属和具有低密度的载体材料的催化剂组合物。应当理解,气凝胶催化剂是通过溶胶-凝胶技术获得的材料,例如从凝胶中真空提取或超临界提取液体,凝胶本身由固体三维网络组成。据信,通过从液体提取到凝胶形成阶段的转变的干燥消除了毛细管力,毛细管力在蒸发中起作用并且导致孔网络的部分或全部坍塌。因此,真空或超临界干燥导致具有低密度、高比表面积、大孔体积和非常多样的孔径的材料。
在一个方面中,载体材料是金属氧化物材料,例如过渡金属氧化物,诸如但不限于二氧化硅、氧化铝、二氧化钛和沸石。在多个方面,载体材料是二氧化硅、氧化铝或沸石。
在另外的方面,氧化铝是γ-al2o3。可选择地,氧化铝可以是热解氧化铝。在还有的另外的方面,沸石是zsm-5、沸石β和usy。
在还有的另外的方面,硅酸盐是热解二氧化硅。在某些方面,载体材料可以是载体材料的组合,诸如二氧化硅和沸石、二氧化硅和氧化铝、二氧化硅和二氧化硅、氧化铝和二氧化硅以及氧化铝和沸石。
用于制备所公开的金属负载的催化剂的方法
根据本公开内容的目的,如本文所体现的和广泛描述的,本公开内容在一个方面涉及用于制备所公开的催化剂的方法。在多个方面,制备所公开的组合物的方法包括初湿含浸技术。在另外的方面,公开了制备所公开的组合物的方法,包括溶胶-凝胶技术。
在多个方面,公开了制备所公开的组合物的方法,该方法包括以下步骤:使载体材料与包含第一3d过渡金属盐和第二3d过渡金属的水溶液接触,以形成包含载体材料以及包含第一3d过渡金属盐和第二3d过渡金属金属的水溶液的混合物;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;并且其中第一3d金属盐和第二3d金属盐不相同;干燥混合物以形成干燥的混合物;煅烧所述干燥的混合物以形成煅烧的混合物;以及还原所述煅烧的混合物,从而提供包含两种3d过渡金属和载体材料的组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自3d过渡金属,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石或其混合物。
在多个方面,公开了制备所公开的组合物的方法,该方法包括以下步骤:使载体材料与包含第一3d过渡金属盐和第二3d过渡金属的水溶液接触,以形成包含载体材料以及包含第一3d过渡金属盐和第二3d过渡金属金属的水溶液的混合物;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;并且其中第一3d金属盐和第二3d金属盐不相同;干燥混合物以形成干燥的混合物;煅烧所述干燥的混合物以形成煅烧的混合物;以及还原所述煅烧的混合物,从而提供包含两种3d过渡金属和载体材料的组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自ni、fe、co、mn、cr、mo及其组合,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石或其混合物。
在多个方面,公开了制备包含3d过渡金属和载体材料的组合物的方法,其中3d过渡金属选自ni、fe、co、mn、cr、mo及其组合;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在;该方法包括以下步骤:使载体材料与包含3d过渡金属盐的水溶液接触,所述3d过渡金属盐选自镍盐、铁盐、钴盐、锰盐、铬盐、钼盐或其组合;形成包含载体材料和包含3d过渡金属盐的水溶液的混合物;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中3d过渡金属盐以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在。
在多个方面,公开了制备包含3d过渡金属和载体材料的组合物的方法,其中3d过渡金属选自ni、fe和co;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在;该方法包括以下步骤:使载体材料与包含3d过渡金属盐的水溶液接触,所述3d过渡金属盐选自镍盐、铁盐和钴盐;形成包含载体材料和包含3d过渡金属盐的水溶液的混合物;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中3d过渡金属盐以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在。
用于制备碳纳米管和氢气的方法
根据本公开内容的目的,如本文所体现的和广泛描述的,本公开内容在一个方面涉及用于在所公开的催化剂的存在下从包括低级烃的反应物气体同时生产碳纳米管和氢气的方法和组合物,所述低级烃包括甲烷、乙烷、丙烷、丁烷或其组合。在多个方面,本公开内容涉及用于使用低级烃作为反应物的伴随生产碳纳米管的无cox的氢气生产方法,所述低级烃包括甲烷、乙烷、丙烷、丁烷或其组合。在多个方面,本公开内容涉及用于利用所公开的催化剂从低级烃同时生产碳纳米管和氢气的方法和组合物,所述低级烃包括甲烷、乙烷、丙烷、丁烷或其组合。
还公开了用于利用所公开的催化剂从低级烃例如甲烷同时生产碳纳米管的高效的无cox的氢气生产方法。在多个方面,所生产的碳纳米管是基底生长的碳纳米管和尖端生长的碳纳米管的混合物。在一些方面,所述方法提供在所公开的催化剂上的选择性的基底生长的碳纳米管。“低级烃”通常是任何含气态烃或挥发性烃的化合物,包括但不限于c1-c4烷烃,例如甲烷、乙烷、丙烷和丁烷。在某些方面,低级烃是甲烷。
还公开了分解低级烃的方法,该方法包括以下步骤:以约1℃min-1至约20℃min-1的加热速率将催化剂床加热到约500℃至约1000℃的温度;其中催化剂床包括所公开的组合物;其中所公开的组合物包含两种3d过渡金属和载体材料,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自3d过渡金属,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中催化剂床被定位在固定流动反应器的反应器床内;其中固定流动反应器包括用于气体流的入口端和出口端;基于约0.1克催化剂负载量,以约25mlmin-1至约200mlmin-1的惰性气体流量通过入口端提供第一惰性气体的流(除非另有指示,否则在本申请中给出的所有气体流量均基于0.1g催化剂负载量);其中第一惰性气体的流与催化剂床接触;终止第一惰性气体的流通过入口端;以等于约5,000h-1至约60,000h-1的空速的反应物气体流量和从约0分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流;其中反应物气体的流与催化剂床接触;并且其中反应物气体包括低级烃和第二惰性气体;在出口处收集流出气体;其中流出气体包括氢气。
还公开了分解低级烃的方法,该方法包括以下步骤:以约1℃min-1至约20℃min-1的加热速率将催化剂床加热到约500℃至约1000℃的温度;其中催化剂床包括所公开的组合物;其中所公开的组合物包含两种3d过渡金属和载体材料,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自ni、fe、co、mn、cr、mo及其组合,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中催化剂床被定位在固定流动反应器的反应器床内;其中固定流动反应器包括用于气体流的入口端和出口端;基于约0.1克催化剂负载量,以约25mlmin-1至约200mlmin-1的惰性气体流量通过入口端提供第一惰性气体的流(除非另有指示,否则在本申请中给出的所有气体流量均基于0.1g催化剂负载量);其中第一惰性气体的流与催化剂床接触;终止第一惰性气体的流通过入口端;以等于约5,000h-1至约60,000h-1的空速的反应物气体流量和从约0分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流;其中反应物气体的流与催化剂床接触;并且其中反应物气体包括低级烃和第二惰性气体;在出口处收集流出气体;其中流出气体包括氢气。
还公开了分解低级烃的方法,该方法包括以下步骤:以约1℃min-1至约20℃min-1的加热速率将催化剂床加热到约500℃至约1000℃的温度;其中催化剂床包括所公开的组合物;其中所公开的组合物包含3d过渡金属和载体材料,其中3d过渡金属选自ni、fe、co、mn、cr、mo及其组合;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中催化剂床被定位在固定流动反应器的反应器床内;其中固定流动反应器包括用于气体流的入口端和出口端;以约25mlmin-1至约200mlmin-1的惰性气体流量通过入口端提供第一惰性气体的流;其中第一惰性气体的流与催化剂床接触;终止第一惰性气体的流通过入口端;以等于约5,000h-1至约60,000h-1的空速的反应物气体流量和从约0分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流;其中反应物气体的流与催化剂床接触;并且其中反应物气体包括低级烃和第二惰性气体;在出口处收集流出气体;其中流出气体包括氢气。
还公开了分解低级烃的方法,该方法包括以下步骤:以约1℃min-1至约20℃min-1的加热速率将催化剂床加热到约500℃至约1000℃的温度;其中催化剂床包括所公开的组合物;其中所公开的组合物包含3d过渡金属和载体材料,其中3d过渡金属选自ni、fe和co;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约40wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约60wt%至约30wt%的量存在;其中载体材料选自二氧化硅、氧化铝、沸石或其混合物;其中催化剂床被定位在固定流动反应器的反应器床内;其中固定流动反应器包括用于气体流的入口端和出口端;以约25mlmin-1至约200mlmin-1的惰性气体流量通过入口端提供第一惰性气体的流;其中第一惰性气体的流与催化剂床接触;终止第一惰性气体的流通过入口端;以约25mlmin-1至约200mlmin-1的反应物气体流量和约20,000h-1至约60,000h-1的空速以及从约0分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流;其中反应物气体的流与催化剂床接触;并且其中反应物气体包括低级烃和第二惰性气体;在出口处收集流出气体;其中流出气体包括氢气。
如本文中所使用的,除非另有说明,否则“碳纳米管”共同地是指多种碳纳米管,包括螺旋碳纳米管、多壁碳纳米管、双壁碳纳米管和单壁碳纳米管。应当理解,除非另有说明,否则被称为碳纳米管的材料或组合物可以包含不同比例的这些子类型的碳纳米管。在一些方面,所提及的碳纳米管基本上包括所有特定的子类型,例如多壁碳纳米管。
如本文中所使用的,“碳材料”是指以可变重量比的碳纳米管、碳纤维、碳纳米颗粒、无定形碳、热解碳和烟灰。
方面
以下示例性方面的列表支持本文提供的公开内容,并且由本文提供的公开内容支持。
方面1.一种包含两种3d过渡金属和载体材料的组合物,其中包含两种3d过渡金属的组合物由式xm:yn表示,其中m和n各自独立地选自3d过渡金属,条件是m和n不相同;其中x和y表示m:n的摩尔比,其中x是具有在约1至约20之间的值的数,y是具有在约1至约10之间的值的数;并且其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物。
方面2.如方面1所述的组合物,其中m和n独立地选自ni、fe、co、mn、cr和mo,条件是m和n不相同。
方面3.如方面1所述的组合物,其中m是ni;并且其中n是fe或co。
方面4.如方面1的组合物,其中m是fe;并且其中n是co。
方面5.如方面1至方面4中任一项所述的组合物,其中x是具有在约1和约10之间的值的数。
方面6.如方面1至方面4中任一项所述的组合物,其中x是具有在约4和约9之间的值的数。
方面7.如方面1至方面4中任一项所述的组合物,其中x是具有约9的值的数。
方面8.如方面1至方面7中任一项所述的组合物,其中y是具有在约1和约5之间的值的数。
方面9.如方面1至方面7中任一项所述的组合物,其中y是具有在约1和约3之间的值的数。
方面10.如方面1至方面7中任一项所述的组合物,其中y是具有约1的值的数。
方面11.如方面1至方面4中任一项所述的组合物,其中x和y是整数。
方面12.如方面11所述的组合物,其中x是具有在1和10之间的整数值的数。
方面13.如方面11所述的组合物,其中x是具有在4和9之间的整数值的数。
方面14.如方面11所述的组合物,其中x是具有9的整数值的数。
方面15.如方面11至方面14中任一项所述的组合物,其中y是具有在1和5之间的整数值的数。
方面16.如方面11至方面14中任一项所述的组合物,其中y是具有在1和3之间的整数值的数。
方面17.如方面11至方面14中任一项所述的组合物,其中y是具有整数值1的数。
方面18.如方面1至方面17中任一项所述的组合物,其中载体材料是沸石。
方面19.如方面18所述的组合物,其中沸石选自zsm-5、沸石β和usy沸石。
方面20.如方面18或方面19所述的组合物,其中沸石具有从约5至约25的sio2/al2o3摩尔比率。
方面21.如方面18或方面19所述的组合物,其中沸石具有从约7.5至约15的sio2/al2o3摩尔比率。
方面22.如方面18或方面19所述的组合物,其中沸石具有从约10至约15的sio2/al2o3摩尔比率。
方面23.如方面18或方面19所述的组合物,其中沸石具有约11.5的sio2/al2o3摩尔比率。
方面24.如方面18至方面28中任一项所述的组合物,其中沸石具有从约250m2·g-1至约600m2·g-1的表面积。
方面25.如方面18至方面28中任一项所述的组合物,其中沸石具有从约350m2·g-1至约500m2·g-1的表面积。
方面26.如方面18至方面28中任一项所述的组合物,其中沸石具有约400m2·g-1至约450m2·g-1的表面积。
方面27.如方面18至方面28中任一项所述的组合物,其中沸石具有约425m2·g-1的表面积。
方面28.如方面1至方面17中任一项所述的组合物,其中载体材料是氧化铝。
方面29.如方面28所述的组合物,其中氧化铝是γ-al2o3或热解氧化铝。
方面30.如方面28所述的组合物,其中氧化铝是γ-al2o3。
方面31.如方面28至方面30中任一项所述的组合物,其中氧化铝的表面积为从约50m2·g-1至约500m2·g-1。
方面32.如方面28至方面30中任一项所述的组合物,其中氧化铝的表面积为从约75m2·g-1至约250m2·g-1。
方面33.如方面28至方面30中任一项所述的组合物,其中氧化铝的表面积为从约100m2·g-1至约200m2·g-1。
方面34.如方面1至方面17中任一项所述的组合物,其中载体材料是二氧化硅。
方面35.如方面34所述的组合物,其中二氧化硅选自沉淀二氧化硅、硅胶(silicagel)和热解二氧化硅。
方面36.如方面34所述的组合物,其中二氧化硅是热解二氧化硅。
方面37.如方面36所述的组合物,其中二氧化硅具有约50kg·m-3至约450kg·m-3的堆积密度(bulkdensity)。
方面38.如方面36所述的组合物,其中二氧化硅具有约90kg·m-3至约380kg·m-3的堆积密度。
方面39.如方面36所述的组合物,其中二氧化硅具有约350kg·m-3至约450kg·m-3的堆积密度。
方面40.如方面36所述的组合物,其中二氧化硅具有约380kg·m-3±约30kg·m-3的堆积密度。
方面41.如方面36至方面40中任一项所述的组合物,其中二氧化硅具有约1nm至约20nm的平均粒度。
方面42.如方面41所述的组合物,其中二氧化硅具有约5nm至约15nm的平均粒度。
方面43.如方面41所述的组合物,其中二氧化硅具有约5nm至约10nm的平均粒度。
方面44.一种制备方面1至方面43中任一项所述的组合物的方法,所述方法包括以下步骤:使载体材料与包含第一3d过渡金属盐和第二3d过渡金属的水溶液接触,以形成包含载体材料以及包含第一3d过渡金属盐和第二3d过渡金属金属的水溶液的混合物;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;并且其中第一3d金属盐和第二3d金属盐不相同;干燥混合物以形成干燥的混合物;煅烧干燥的混合物以形成煅烧的混合物;以及还原所煅烧的混合物,从而提供方面1至方面43中任一项所述的组合物。
方面45.如方面44所述的方法,其中载体材料是二氧化硅。
方面46.如方面45所述的方法,其中二氧化硅选自沉淀二氧化硅、硅胶和热解二氧化硅。
方面47.如方面45所述的方法,其中二氧化硅是热解二氧化硅。
方面48.如方面47所述的方法,其中二氧化硅具有约50kg·m-3至约450kg·m-3的堆积密度。
方面49.如方面47所述的方法,其中二氧化硅具有约90kg·m-3至约380kg·m-3的堆积密度。
方面50.如方面47所述的方法,其中二氧化硅具有约350kg·m-3至约450kg·m-3的堆积密度。
方面51.如方面47所述的方法,其中二氧化硅具有约380kg·m-3±约30kg·m-3的堆积密度。
方面52.如方面47至方面51中任一项所述的方法,其中二氧化硅具有约1nm至约20nm的平均粒度。
方面53.如方面52所述的方法,其中二氧化硅具有约5nm至约15nm的平均粒度。
方面54.如方面52所述的方法,其中二氧化硅具有约5nm至约10nm的平均粒度。
方面55.如方面44至方面54中任一项所述的方法,其中第一3d金属盐和第二3d金属盐独立地选自镍盐、铁盐、钴盐、钼盐、铬盐、锰盐及其组合;条件是第一3d金属盐和第二3d金属盐不相同。
方面56.如方面55所述的方法,第一3d金属盐是镍盐并且第二3d金属盐是铁盐或钴盐。
方面57.如方面55所述的方法,第一3d金属盐是铁盐并且第二3d金属盐是钴盐。
方面58.如方面55至方面57中任一项所述的方法,其中镍盐是ni(no3)2·6h2o。
方面59.如方面55至方面57中任一项所述的方法,其中铁盐是fe(no3)2·9h2o。
方面60.如方面55至方面57中任一项所述的方法,其中钴盐是co(no3)2·6h2o。
方面61.如方面44至方面60中任一项所述的方法,其中干燥所述混合物包括在大于100℃至约200℃的温度加热混合物。
方面62.如方面44至方面60中任一项所述的方法,其中干燥所述混合物包括在约120℃至约150℃的温度加热混合物。
方面63.如方面44至方面60中任一项所述的方法,其中干燥所述混合物包括在约120℃至约140℃的温度加热混合物。
方面64.如方面44至方面63中任一项所述的方法,其中干燥包括在烘箱中加热混合物。
方面65.如方面64所述的方法,其中干燥包括在烘箱中加热持续约4小时至约36小时的时间段。
方面66.如方面64所述的方法,其中干燥包括在烘箱中加热持续约12小时至约24小时的时间段。
方面67.如方面44至方面66中任一项所述的方法,其中煅烧在约400℃至约900℃的温度发生,其中加热速率为约1℃·min-1至约10℃·min-1。
方面68.如方面67所述的方法,其中当第一3d金属盐是镍盐并且第二3d金属盐是铁盐时,温度为约450℃至约550℃并且加热速率为约3℃·min-1至约7℃·min-1。
方面69.如方面68所述的方法,其中温度为约500℃并且加热速率为约5℃·min-1。
方面70.如方面67所述的方法,其中当第一3d金属盐是镍盐并且第二3d金属盐是钴盐时,温度为约700℃至约800℃并且加热速率为约3℃·min-1至约7℃·min-1。
方面71.如方面70所述的方法,其中温度为约750℃并且加热速率为约5℃·min-1。
方面72.如方面67所述的方法,其中当第一3d金属盐是铁盐并且第二3d金属盐是钴盐时,温度为约400℃至约500℃并且加热速率为约3℃·min-1至约7℃·min-1。
方面73.如方面72所述的方法,其中温度为约450℃并且加热速率为约5℃·min-1。
方面74.如方面44至方面73中任一项所述的方法,其中煅烧在马弗炉中发生。
方面75.如方面44至方面74中任一项所述的方法,其中通过以约50ml·min-1至约150ml·min-1的流量提供还原气体混合物并且在约400℃至约900℃的温度以约1℃至约20℃的加热速率加热持续约1小时至约12小时来进行还原;其中还原气体混合物包括约5%至约20%的氢气和约95%至约80%的氩气。
方面76.如方面75所述的方法,其中流量为约50ml·min-1至约100ml·min-1;并且其中还原气体混合物包括约5%至约15%的氢气。
方面77.如方面75所述的方法,其中流量为约70ml·min-1;并且其中还原气体混合物包括约10%的氢气。
方面78.如方面44至方面77中任一项所述的方法,其中当第一3d金属盐是镍盐并且第二3d金属盐是铁盐时,在约650℃至约750℃的温度发生还原并且加热速率为约7℃·min-1至约12℃·min-1持续约3小时至约5小时。
方面79.如方面62所述的方法,其中温度为约700℃并且加热速率为约10℃·min-1持续约4小时。
方面80.如方面44至方面77中任一项所述的方法,其中当第一3d金属盐是镍盐并且第二3d金属盐是钴盐时,在约650℃至约750℃的温度发生还原并且加热速率为约7℃·min-1至约12℃·min-1持续约3小时至约3小时。
方面81.如方面80所述的方法,其中温度为约700℃并且加热速率为约10℃·min-1持续约2小时。
方面82.如方面44至方面77中任一项所述的方法,其中当第一3d金属盐是铁盐并且第二3d金属盐是钴盐时,在约450℃至约550℃的温度发生还原并且加热速率为约7℃·min-1至约12℃·min-1持续约3小时至约5小时。
方面83.如方面80所述的方法,其中温度为约580℃并且加热速率为约10℃·min-1持续约4小时。
方面84.一种组合物,其由方面44至方面83中任一项所述的方法制备。
方面85.一种分解低级烃的方法,所述方法包括以下步骤:以约1℃·min-1至约20℃·min-1的加热速率将催化剂加热到约500℃至约1000℃的温度;其中所述催化剂床包括方面1至方面43中任一项所述的组合物或通过方面44至方面83中任一项所述的方法制备的组合物;其中催化剂被定位在反应器内;其中反应器是固定床反应器或移动床流动反应器构造;其中反应器包括用于气体流的入口端和出口端;任选地(a)以约25ml·min-1至约200ml·min-1的惰性气体流量通过入口端提供第一惰性气体的流;其中第一惰性气体的流与催化剂接触;以及(b)终止第一惰性气体的流通过入口端;以等于约5,000h-1至约60,000h-1的空速的反应物气体流和从约0分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流;其中反应物气体的流与催化剂床接触;并且其中反应物气体包括低级烃和第二惰性气体;在出口处收集流出气体;其中所述流出气体包括氢气。
方面86.如方面85所述的方法,其中第一惰性气体包括氮气、氩气或其混合物。
方面87.如方面85所述的方法,其中第一惰性气体包括氮气。
方面88.如方面85至方面87中任一项所述的方法,其中惰性气体流量为约50ml·min-1至约150ml·min-1。
方面89.如方面85至方面87中任一项所述的方法,其中惰性气体流量为约50ml·min-1至约100ml·min-1。
方面90.如方面85至方面87中任一项所述的方法,其中惰性气体流量为约60ml·min-1至约80ml·min-1。
方面91.如方面85至方面87中任一项所述的方法,其中惰性气体流量为约70ml·min-1。
方面92.如方面85至方面91中任一项所述的方法,其中反应物气体包括从约10%至约60%的低级烃和从约90%至约40%的第二惰性气体。
方面93.如方面92所述的方法,其中反应物气体包括从约20%至约50%的低级烃和从约80%至约50%的第二惰性气体。
方面94.如方面85至方面93中任一项所述的方法,其中低级烃是甲烷。
方面95.如方面85至方面94中任一项所述的方法,其中第二惰性气体选自氮气、氩气或其混合物。
方面96.如方面85至方面95中任一项所述的方法,其中第二惰性气是氮气。
方面97.如方面85至方面96中任一项所述的方法,其中反应物气体流量为约50ml·min-1至约150ml·min-1。
方面98.如方面85至方面96中任一项所述的方法,其中反应物气体流量为约50ml·min-1至约100ml·min-1。
方面99.如方面85至方面96中任一项所述的方法,其中反应物气体流量为约60ml·min-1至约80ml·min-1。
方面100.如方面85至方面96中任一项所述的方法,其中反应物气体流量为约70ml·min-1。
方面101.如方面85至方面100中任一项所述的方法,其中空速为约35,000h-1至约50,000h-1。
方面102.如方面85至方面100中任一项所述的方法,其中空速为约40,000h-1至约45,000h-1。
方面103.如方面85至方面100中任一项所述的方法,其中空速为约42,000h-1。
方面104.如方面85至方面103中任一项所述的方法,其中运行时间为从约0分钟至约90分钟。
方面105.如方面85至方面103中任一项所述的方法,其中运行时间为从约0分钟至约75分钟。
方面106.如方面85至方面103中任一项所述的方法,其中运行时间为从约0分钟至约60分钟。
方面107.如方面85至方面106中任一项所述的方法,其中低级烃的分解以约10%至约90%的转化效率产生氢气。
方面108.如方面85至方面106中任一项所述的方法,其中低级烃的分解以约30%至约90%的转化效率产生氢气。
方面109.如方面85至方面106中任一项所述的方法,其中低级烃的分解以至少约35%至至少约70%的转化效率产生氢气。
方面110.如方面85至方面109中任一项所述的方法,其中低级烃的分解产生碳。
方面111.如方面110所述的方法,其中碳基本上不含无定形碳。
方面112.如方面90或方面111所述的方法,其中碳在与催化剂床接触时积聚。
方面113.如方面110至方面112中任一项所述的方法,其中碳包括碳纳米管。
方面114.如方面113所述的方法,其中碳纳米管具有约0.600至约0.880的id/ig值。
方面115.如方面113所述的方法,其中碳纳米管具有约0.760至约0.875的id/ig值。
方面116.如方面113至方面115中任一项所述的方法,其中碳纳米管包括多壁碳纳米管。
方面117.如方面113至方面116中任一项所述的方法,其中碳纳米管具有约1nm至约150nm的平均直径。
方面118.如方面113至方面116中任一项所述的方法,其中碳纳米管具有约5nm至约70nm的平均直径。
方面119.如方面113至方面116中任一项所述的方法,其中碳纳米管具有约5nm至约30nm的平均直径。
方面120.如方面113至方面116中任一项所述的方法,其中碳纳米管具有约40nm至约150nm的平均直径。
方面121.如方面113至方面116中任一项所述的方法,其中碳纳米管具有约40nm至约70nm的平均直径。
方面122.如方面113至方面116中任一项所述的方法,其中碳纳米管具有约100nm至约140nm的平均直径。
方面123.如方面113至方面122中任一项所述的方法,其中碳纳米管包括相对于碳纳米管的管状轴线倾斜堆叠的石墨烯层。
方面124.如方面113至方面122中任一项所述的方法,其中碳纳米管包括相对于碳纳米管的管状轴线平行堆叠的石墨烯层。
方面125.如方面113至方面124中任一项所述的方法,其中碳纳米管包括尖端生长的碳纳米管、基底生长的碳纳米管及其混合物。
方面126.如方面113至方面125中任一项所述的方法,还包括从催化剂床纯化碳纳米管。
方面127.如方面113至方面126中任一项所述的方法,还包括再生循环,所述再生循环包括终止反应物气体的流并向催化剂床提供再生气体;其中再生气体与催化剂床接触持续从约5分钟至约240分钟的接触时间段;其中催化剂床的温度为从约250℃至约750℃;并且其中再生气体包括氧气。
方面128.如方面127所述的方法,其中再生气体包括从约5%至约25%的氧气。
方面129.如方面127所述的方法,其中再生气体包括从约7.5%至约15%的氧气。
方面130.如方面127所述的方法,其中再生气体包括约10%的氧气。
方面131.如方面127至方面130中任一项所述的方法,其中接触时间段为从约15分钟至约60分钟。
方面132.如方面127至方面130中任一项所述的方法,其中接触时间段为从约20分钟至约40分钟。
方面133.如方面127至方面130中任一项所述的方法,其中接触时间段为约30分钟。
方面134.如方面127至方面133中任一项所述的方法,其中催化剂床处于从约400℃至约600℃的温度。
方面135.如方面127至方面133中任一项所述的方法,其中催化剂床处于从约450℃至约550℃的温度。
方面136.如方面127至方面135中任一项所述的方法,其中催化剂床处于约500℃的温度。
方面137.如方面127至方面136中任一项所述的方法,还包括另外的反应循环,所述另外的反应循环包括终止催化剂床与再生气体的接触;任选地以约25ml·min-1至约200ml·min-1的惰性气体流量通过入口端提供第一惰性气体的流;其中第一惰性气体的流与催化剂床接触;终止第一惰性气体的流通过入口端;以约25ml·min-1的反应物气体流量到约20,000h-1至约60,000h-1的空速以及从约10分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流。
方面138.如方面314所述的方法,其中对于每个另外的反应循环,低级烃转化的效率变化不超过10%。
方面139.如方面304至方面314中任一项所述的方法,还包括重复再生循环和另外的反应循环持续1个至10个循环。
方面140.一种碳纳米管,其由方面85至方面139中任一项所述的方法制备。
方面141.一种组合物,包含3d过渡金属和载体材料,
其中3d过渡金属选自ni、fe、co、mn、cr和mo;
其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛及其混合物;
其中3d过渡金属以基于3d过渡金属和载体材料的总重量的从约5wt%至约70wt%的量存在;并且
其中载体材料以基于3d过渡金属和载体材料的总重量的从约95wt%至约30wt%的量存在。
方面142.如方面141所述的组合物,其中载体材料是沸石。
方面143.如方面142所述的组合物,其中沸石选自zsm-5、沸石β和usy沸石。
方面144.如方面142或方面143所述的组合物,其中沸石具有从约5至约25的sio2/al2o3摩尔比率。
方面145.如方面142或方面143所述的组合物,其中沸石具有从约7.5至约15的sio2/al2o3摩尔比率。
方面146.如方面142或方面143所述的组合物,其中沸石具有从约10至约15的sio2/al2o3摩尔比率。
方面147.如方面142或方面143所述的组合物,其中沸石具有约11.5的sio2/al2o3摩尔比率。
方面148.如方面142至方面147中任一项所述的组合物,其中沸石具有从约250m2·g-1至约600m2·g-1的表面积。
方面149.如方面142至方面147中任一项所述的组合物,其中沸石具有从约350m2·g-1至约500m2·g-1的表面积。
方面150.如方面142至方面147中任一项所述的组合物,其中沸石具有约400m2·g-1至约450m2·g-1的表面积。
方面151.如方面142至方面147中任一项所述的组合物,其中沸石具有约425m2·g-1的表面积。
方面152.如方面141所述的组合物,其中载体材料是氧化铝。
方面153.如方面152所述的组合物,其中氧化铝是γ-al2o3或热解氧化铝。
方面154.如方面152所述的组合物,其中氧化铝是γ-al2o3。
方面155.如方面141或方面152至方面154中任一项所述的组合物,其中氧化铝的表面积为从约50m2·g-1至约500m2·g-1。
方面156.如方面141或方面152至方面154中任一项所述的组合物,其中氧化铝的表面积为从约75m2·g-1至约250m2·g-1。
方面157.如方面141或方面152至方面154中任一项所述的组合物,其中氧化铝的表面积为从约100m2·g-1至约200m2·g-1。
方面158.如方面141所述的组合物,其中载体材料是二氧化硅。
方面159.如方面158所述的组合物,其中二氧化硅选自沉淀二氧化硅、硅胶和热解二氧化硅。
方面160.如方面159所述的组合物,其中二氧化硅是热解二氧化硅。
方面161.如方面160所述的组合物,其中二氧化硅具有约50kg·m-3至约450kg·m-3的堆积密度。
方面162.如方面160所述的组合物,其中二氧化硅具有约90kg·m-3至约380kg·m-3的堆积密度。
方面163.如方面160所述的组合物,其中二氧化硅具有约350kg·m-3至约450kg·m-3的堆积密度。
方面164.如方面160所述的组合物,其中二氧化硅具有约380kg·m-3±约30kg·m-3的堆积密度。
方面165.如方面141至方面164中任一项所述的组合物,其中二氧化硅具有约1nm至约200nm的平均粒度。
方面166.如方面165所述的组合物,其中二氧化硅具有约5nm至约150nm的平均粒度。
方面167.如方面165所述的组合物,其中二氧化硅具有约5nm至约100nm的平均粒度。
方面168.如方面165所述的组合物,其中二氧化硅具有约5nm至约50nm的平均粒度。
方面169.如方面165所述的组合物,其中二氧化硅具有约5nm至约40nm的平均粒度。
方面170.如方面165所述的组合物,其中二氧化硅具有约5nm至约30nm的平均粒度。
方面171.如方面165所述的组合物,其中二氧化硅具有约5nm至约20nm的平均粒度。
方面172.如方面165所述的组合物,其中二氧化硅具有约5nm至约15nm的平均粒度。
方面173.如方面165所述的组合物,其中二氧化硅具有约5nm至约10nm的平均粒度。
方面174.如方面141至方面173中任一项所述的组合物,其中组合物包含约55%至约65%的fe和约45%至约35%的载体材料。
方面175.如方面141至方面173中任一项所述的组合物,其中组合物包含约5%至约65%的ni和约95%至约35%的载体材料。
方面176.如方面141至方面173中任一项所述的组合物,其中组合物包含约25%至约65%的co和约75%至约35%的载体材料。
方面177.如方面141至方面173中任一项所述的组合物,其中组合物包含约35%至约65%的ni和约65%至约35%的载体材料。
方面178.如方面141至方面173中任一项的组合物,其中组合物包含约35%至约65%的co和约65%至约35%的载体材料。
方面179.如方面141至方面178中任一项的组合物,其中组合物具有气凝胶结构。
方面180.如方面141至方面179中任一项所述的组合物,其中组合物具有介孔结构。
方面181.如方面141至方面180中任一项所述的组合物,其中组合物具有从约50m2·g-1至约500m2·g-1的bet表面积。
方面182.如方面181所述的组合物,其中组合物具有从约100m2·g-1至约400m2·g-1的bet表面积。
方面183.如方面181所述的组合物,其中组合物具有从约150m2·g-1至约350m2·g-1的bet表面积。
方面184.如方面181所述的组合物,其中组合物具有从约220m2·g-1至约380m2·g-1的bet表面积。
方面185.如方面181所述的组合物,其中组合物具有从约230m2·g-1至约370m2·g-1的bet表面积。
方面186.如方面181所述的组合物,其中组合物具有从约240m2·g-1至约360m2·g-1的bet表面积。
方面187.如方面181所述的组合物,其中组合物具有从约250m2·g-1至约350m2·g-1的bet表面积。
方面188.如方面181所述的组合物,其中组合物具有从约260m2·g-1至约340m2·g-1的bet表面积。
方面189.如方面181所述的组合物,其中组合物具有从约265m2·g-1至约335m2·g-1的bet表面积。
方面190.如方面141至方面180中任一项所述的组合物,其中组合物具有从约1nm至约50nm的barrett-joyner-halenda(bjh)孔径。
方面191.如方面190所述的组合物,其中组合物具有从约5nm至约45nm的barrett-joyner-halenda(bjh)孔径。
方面192.如方面190所述的组合物,其中组合物具有从约5nm至约40nm的barrett-joyner-halenda(bjh)孔径。
方面193.如方面190所述的组合物,其中组合物具有从约5nm至约35nm的barrett-joyner-halenda(bjh)孔径。
方面194.如方面190所述的组合物,其中组合物具有从约5nm至约30nm的barrett-joyner-halenda(bjh)孔径。
方面195.如方面190所述的组合物,其中组合物具有从约5nm至约25nm的barrett-joyner-halenda(bjh)孔径。
方面196.如方面190所述的组合物,其中组合物具有从约5nm至约20nm的barrett-joyner-halenda(bjh)孔径。
方面197.如方面190所述的组合物,其中组合物具有从约5nm至约15nm的barrett-joyner-halenda(bjh)孔径。
方面198.如方面190所述的组合物,其中组合物具有从约5nm至约10nm的barrett-joyner-halenda(bjh)孔径。
方面199.如方面190所述的组合物,其中组合物具有从约10nm至约50nm的barrett-joyner-halenda(bjh)孔径。
方面200.如方面190所述的组合物,其中组合物具有从约10nm至约45nm的barrett-joyner-halenda(bjh)孔径。
方面201.如方面190所述的组合物,其中组合物具有从约10nm至约40nm的barrett-joyner-halenda(bjh)孔径。
方面202.如方面190所述的组合物,其中组合物具有从约10nm至约35nm的barrett-joyner-halenda(bjh)孔径。
方面203.如方面190所述的组合物,其中组合物具有从约10nm至约30nm的barrett-joyner-halenda(bjh)孔径。
方面204.如方面190所述的组合物,其中组合物具有从约10nm至约25nm的barrett-joyner-halenda(bjh)孔径。
方面205.如方面190所述的组合物,其中组合物具有从约10nm至约20nm的barrett-joyner-halenda(bjh)孔径。
方面206.如方面190所述的组合物,其中组合物具有从约10nm至约15nm的barrett-joyner-halenda(bjh)孔径。
方面207.如方面141至方面206中任一项所述的组合物,其中组合物具有从约0.3cm3/g至约1.6cm3/g的孔体积。
方面208.如方面207所述的组合物,其中组合物具有从约0.4cm3/g至约1.4cm3/g的孔体积。
方面209.如方面207所述的组合物,其中组合物具有从约0.5cm3/g至约1.4cm3/g的孔体积。
方面210.如方面207所述的组合物,其中组合物具有从约0.6cm3/g至约1.4cm3/g的孔体积。
方面211.如方面207所述的组合物,其中组合物具有从约0.7cm3/g至约1.4cm3/g的孔体积。
方面212.如方面207所述的组合物,其中组合物具有从约0.8cm3/g至约1.4cm3/g的孔体积。
方面213.如方面207所述的组合物,其中组合物具有从约0.9cm3/g至约1.4cm3/g的孔体积。
方面214.如方面207所述的组合物,其中组合物具有从约1.0cm3/g至约1.4cm3/g的孔体积。
方面215.如方面207所述的组合物,其中组合物具有从约0.3cm3/g至约1.2cm3/g的孔体积。
方面216.如方面207所述的组合物,其中组合物具有从约0.4cm3/g至约1.2cm3/g的孔体积。
方面217.如方面207所述的组合物,其中组合物具有从约0.5cm3/g至约1.2cm3/g的孔体积。
方面218.如方面207所述的组合物,其中组合物具有从约0.6cm3/g至约1.2cm3/g的孔体积。
方面219.如方面207所述的组合物,其中组合物具有从约0.7cm3/g至约1.2cm3/g的孔体积。
方面220.如方面207所述的组合物,其中组合物具有从约0.8cm3/g至约1.2cm3/g的孔体积。
方面221.如方面207所述所述的组合物,其中组合物具有从约0.9cm3/g至约1.2cm3/g的孔体积。
方面222.如方面207所述的组合物,其中组合物具有从约1.0cm3/g至约1.2cm3/g的孔体积。
方面223.方面141至方面222中任一项所述的组合物,其中3d过渡金属被分布在载体材料内、载体材料的外表面上、载体材料的内表面上或其组合。
方面224.一种制备方面141至方面223中任一项所述的组合物的方法,所述方法包括以下步骤:使载体材料与包含选自镍盐、铁盐、钴盐、锰盐、铬盐、钼盐或其组合的3d过渡金属盐的水溶液接触,从而形成包含载体材料和包含3d过渡金属盐的水溶液的混合物;其中载体材料选自二氧化硅、氧化铝、沸石、二氧化钛或其混合物;其中3d过渡金属盐以基于3d过渡金属和载体材料的总重量的从约5wt%至约70wt%的量存在;并且其中载体材料以基于3d过渡金属和载体材料的总重量的从约95wt%至约30wt%的量存在;干燥混合物以形成干燥的混合物;煅烧干燥的混合物以形成煅烧的混合物;以及还原煅烧的混合物,从而提供方面141至方面223中任一项所述的组合物。
方面225.如方面224所述的方法,其中3d过渡金属盐是铁盐。
方面226.如方面224所述的方法,其中铁盐是fe(no3)2·9h2o。
方面227.一种组合物,其由方面224至方面226中任一项所述的方法制备。
方面228.一种制备方面141至方面223中任一项所述的组合物的方法,所述方法包括以下步骤:提供气凝胶前体溶液,所述气凝胶前体溶液包含选自镍盐、铁盐、钴盐、锰盐、铬盐、钼盐或其组合的至少一种3d过渡金属盐;气凝胶载体盐,其选自铝盐、钛盐、硅盐或其组合;水;以及有机溶剂;加热所述气凝胶前体溶液;将所述气凝胶前体与胶凝剂混合,从而形成气凝胶溶胶-凝胶混合物;干燥所述气凝胶溶胶-凝胶混合物;并且煅烧以形成煅烧的气凝胶溶胶-凝胶混合物;还原所煅烧的气凝胶溶胶-凝胶混合物,从而提供方面141至方面223中任一项所述的组合物。
方面229.如方面228所述的方法,其中3d过渡金属盐是镍盐。
方面230.如方面229所述的方法,其中镍盐包括卤化镍盐、硝酸镍盐、硫酸镍盐或其组合。
方面231.如方面229或方面230所述的方法,其中镍盐包括ni(ii)。
方面232.如方面228所述的方法,其中3d过渡金属盐是钴盐。
方面233.如方面232所述的方法,其中钴盐包括卤化钴盐、硝酸钴盐、硫酸钴盐或其组合。
方面234.如方面232或方面233所述的方法,其中钴盐包括钴(ii)。
方面235.如方面228至方面234中任一项所述的方法,其中气凝胶载体盐是铝盐。
方面236.如方面229所述的方法,其中铝盐包括氯化铝盐。
方面237.如方面236所述的方法,其中铝盐包括alcl3。
方面238.如方面228至方面237中任一项所述的方法,其中有机溶剂是c1-c6醇。
方面239.如方面238所述的方法,其中有机溶剂是甲醇、乙醇、丙醇或其组合。
方面240.如方面228至方面239中任一项所述的方法,其中胶凝剂是环氧丙烷。
方面241.如方面228至方面240中任一项所述的方法,其中煅烧包括在气凝胶煅烧温度加热持续气凝胶煅烧时间段;其中气凝胶煅烧温度为从约300℃至约1000℃;并且其中气凝胶煅烧时间段为从约1分钟至约48小时。
方面242.如方面241所述的方法,其中气凝胶煅烧温度为从约300℃至约900℃。
方面243.如方面241所述的方法,其中气凝胶煅烧温度为从约300℃至约800℃。
方面244.如方面241所述的方法,其中气凝胶煅烧温度为从约300℃至约700℃。
方面245.如方面241所述的方法,其中气凝胶煅烧温度为从约300℃至约600℃。
方面246.如方面241所述的方法,其中气凝胶煅烧温度为从约300℃至约500℃。
方面247.如方面241所述的方法,其中气凝胶煅烧温度为从约400℃至约1000℃。
方面248.如方面241所述的方法,其中气凝胶煅烧温度为从约400℃至约900℃。
方面249.如方面241所述的方法,其中气凝胶煅烧温度为从约400℃至约800℃。
方面250.如方面241所述的方法,其中气凝胶煅烧温度为从约400℃至约700℃。
方面251.如方面241所述的方法,其中气凝胶煅烧温度为从约400℃至约600℃。
方面252.如方面241所述的方法,其中气凝胶煅烧温度为从约400℃至约500℃。
方面253.如方面241至方面252中任一项所述的方法,其中气凝胶煅烧时间段为从约30分钟至约24小时。
方面254.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约18小时。
方面255.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约16小时。
方面256.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约14小时。
方面257.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约12小时。
方面258.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约10小时。
方面259.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约9小时。
方面260.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约8小时。
方面261.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约7小时。
方面262.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约6小时。
方面263.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约5小时。
方面264.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约4小时。
方面265.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约3小时。
方面266.如方面253所述的方法,其中气凝胶煅烧时间段为从约1小时至约2小时。
方面267.一种组合物,其由方面228至方面266中任一项所述的方法制备。
方面268.一种分解低级烃的方法,所述方法包括以下步骤:以约1℃·min-1至约20℃·min-1的加热速率将催化剂床加热到约500℃至约1000℃的温度;其中催化剂床包括方面141至方面223中任一项所述的组合物,由方面224至方面226中任一项所述的方法制备的组合物;由方面228至方面266中任一项所述的方法制备的组合物;其中催化剂被定位在反应器内;其中反应器是固定床或移动床流动反应器构造;其中反应器包括用于气体流的入口端和出口端;任选地:(a)以约25ml·min-1至约200ml·min-1的惰性气体流量通过入口端提供第一惰性气体的流;其中第一惰性气体的流与催化剂床接触;以及(b)终止第一惰性气体的流通过入口端,如果存在的话;以约25ml·min-1的反应物气体流量到约5,000h-1至约60,000h-1的空速以及从约0分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流;其中反应物气体的流与催化剂接触;并且其中反应物气体包括低级烃和第二惰性气体;在出口处收集流出气体;其中所述流出气体包括氢气。
方面269.如方面268所述的方法,其中第一惰性气体包括氮气、氩气或其混合物。
方面270.如方面268所述的方法,其中第一惰性气体包括氮气。
方面271.如方面268至方面271中任一项所述的方法,其中惰性气体流量为约50ml·min-1至约150ml·min-1。
方面272.如方面268至方面271中任一项所述的方法,其中惰性气体流量为约50ml·min-1至约100ml·min-1。
方面273.如方面268至方面271中任一项所述的方法,其中惰性气体流量为约60ml·min-1至约80ml·min-1。
方面274.如方面268至方面271中任一项所述的方法,其中惰性气体流量为约70ml·min-1。
方面275.如方面268至方面274中任一项所述的方法,其中反应物气体包括从约10%至约60%的低级烃和从约90%至约40%的第二惰性气体。
方面276.如方面275所述的方法,其中反应物气体包括从约20%至约50%的低级烃和从约80%至约50%的第二惰性气体。
方面277.如方面268至方面276中任一项所述的方法,其中低级烃是甲烷。
方面278.如方面268至方面277中任一项所述的方法,其中第二惰性气体选自氮气、氩气或其混合物。
方面279.如方面268至方面277中任一项所述的方法,其中第二惰性气是氮气。
方面280.如方面268至方面279中任一项所述的方法,其中反应物气体流量为约50ml·min-1至约150ml·min-1。
方面281.如方面268至方面279中任一项所述的方法,其中反应物气体流量为约50ml·min-1至约100ml·min-1。
方面282.如方面268至方面279中任一项所述的方法,其中反应物气体流量为约60ml·min-1至约80ml·min-1。
方面283.如方面268至方面279中任一项所述的方法,其中反应物气体流量为约70ml·min-1。
方面284.如方面268至方面283中任一项所述的方法,其中空速为约35,000h-1至约50,000h-1。
方面285.如方面268至方面283中任一项所述的方法,其中空速为约40,000h-1至约45,000h-1。
方面286.如方面268至方面283中任一项所述的方法,其中空速为约42,000h-1。
方面287.如方面268至方面286中任一项所述的方法,其中运行时间为从约0分钟至约90分钟。
方面288.如方面268至方面103中任一项所述的方法,其中运行时间为从约0分钟至约75分钟。
方面289.如方面268至方面103中任一项所述的方法,其中运行时间为从约0分钟至约60分钟。
方面290.如方面268至方面106中任一项所述的方法,其中低级烃的分解以约10%至约90%的转化效率产生氢气。
方面291.如方面268至方面106中任一项所述的方法,其中低级烃的分解以约30%至约90%的转化效率产生氢气。
方面292.如方面268至方面106中任一项所述的方法,其中低级烃的分解以至少约35%至至少约70%的转化效率产生氢气。
方面293.如方面268至方面292中任一项所述的方法,其中催化剂床包括约55%至约65%的fe和约45%至约35%的载体材料的组合物。
方面294.如方面268至方面293中任一项所述的方法,其中低级烃包括甲烷。
方面295.如方面268至方面294中任一项所述的方法,其中低级烃的分解产生碳材料。
方面296.如方面295所述的方法,其中碳材料基本上不含无定形碳。
方面297.如方面295或方面296所述的方法,其中碳在与催化剂床接触时积聚。
方面298.如方面295至方面297中任一项所述的方法,其中碳包括碳纳米管。
方面299.如方面298所述的方法,其中碳纳米管具有约40nm至约150nm的平均直径。
方面300.如方面298所述的方法,其中碳纳米管具有约40nm至约70nm的平均直径。
方面301.如方面298所述的方法,其中碳纳米管具有约100nm至约140nm的平均直径。
方面302.如方面298至方面301中任一项所述的方法,其中碳纳米管包括尖端生长的碳纳米管、基底生长的碳纳米管及其混合物。
方面303.如方面302所述的方法,其中大于50%的碳纳米管包括基底生长的碳纳米管。
方面304.如方面268至方面303中任一项所述的方法,还包括再生循环,所述再生循环包括终止反应物气体的流并向催化剂床提供再生气体;其中再生气体与催化剂床接触持续从约5分钟至约240分钟的接触时间段;其中催化剂床的温度为从约250℃至约750℃;并且其中再生气体包括氧气。
方面305.如方面304所述的方法,其中再生气体包括从约5%至约25%的氧气。
方面306.如方面304所述的方法,其中再生气体包括从约7.5%至约15%的氧气。
方面307.如方面304所述的方法,其中再生气体包括约10%的氧气。
方面308.如方面304至方面307中任一项所述的方法,其中接触时间段为从约15分钟至约60分钟。
方面309.如方面304至方面307中任一项所述的方法,其中接触时间段为从约20分钟至约40分钟。
方面310.如方面304至方面307中任一项所述的方法,其中接触时间段为约30分钟。
方面311.如方面304至方面310中任一项所述的方法,其中催化剂床处于从约400℃至约600℃的温度。
方面312.如方面304至方面310中任一项所述的方法,其中催化剂床处于从约450℃至约550℃的温度。
方面313.如方面304至方面310中任一项所述的方法,其中催化剂床处于500℃的温度。
方面314.如方面304至方面313中任一项所述的方法,还包括另外的反应循环,所述另外的反应循环包括终止催化剂床与再生气体的接触;任选地以约25ml·min-1至约200ml·min-1的惰性气体流量通过入口端提供第一惰性气体的流;其中第一惰性气体的流与催化剂床接触;终止第一惰性气体的流通过入口端;以约25ml·min-1的反应物气体流量到约20,000h-1至约60,000h-1的空速以及从约10分钟至约240分钟的运行时间(tos)提供通到入口端的反应物气体的流。
方面315.如方面314所述的方法,其中对于另外的反应循环,低级烃转化的效率变化不超过10%。
方面316.如方面304至方面314中任一项所述的方法,还包括重复再生循环和另外的反应循环持续1个至10个循环。
方面317.一种碳纳米管,其由方面268至方面316中任一项所述的方法制备。
方面318.一种用于生产多壁碳纳米管的方法,包括以下步骤:在基于金属的催化剂的存在下分解甲烷,其中基于金属的催化剂至少再生一次;生长具有尖端和基底的多壁碳纳米管,其中纳米管的基底被附接至基于金属的催化剂并从基底向外生长;以及将生长的多壁碳纳米管与生长的纳米管的基底附近的基于金属的催化剂分离,留下基于金属的催化剂以生长新的纳米管。
方面319.如方面318所述的方法,还包括生产基本上无cox的氢气,作为生产多壁碳纳米管的副产物。
方面320.一种碳纳米管,其由方面318或方面319所述的方法制备。
从上文中将看出,本文的方面很好地适用于实现上文阐述的所有目的和目标,以及明显的并且结构所固有的其他优点。
虽然结合彼此讨论了特定的元件和步骤,但是应当理解,本文提供的任何元件和/或步骤都被预期为与任何其他元件和/或步骤可组合,而不管它们的明确规定,同时仍然在本文提供的范围内。
将理解的是,某些特征和子组合具有实用性并且可以在不参考其他特征和子组合的情况下使用。这由权利要求所预期并且在权利要求的范围内。
因为在不脱离本发明的范围的情况下可以做出许多可能的方面,所以应当理解,在本文阐述或在附图和详细描述中示出的所有内容应当被解释为说明性的,并且非限制性的意义。
还应当理解,本文中使用的术语仅仅是为了描述特定方面的目的,并且不意图是限制性的。本领域技术人员将认识到本文描述的方面的许多变型和修改。这些变型和修改意图被包括在本公开内容的教导中,并且意图被本文的权利要求所涵盖。
现在已经描述了本公开内容的方面,通常,以下实施例描述了本公开内容的一些另外的方面。虽然结合以下实施例和对应的文字以及附图来描述本公开内容的方面,但不意图将本公开内容的方面限于该描述。相反,意图覆盖包括在本公开内容的精神和范围内的所有替代方案、修改和等效物。
实施例
提出以下实施例以便向本领域普通技术人员提供如何制备和评估本文要求保护的化合物、组合物、物品、装置和/或方法的完全公开内容和描述,并且意图是本公开内容的单纯实例性的,并且不意图限制发明人视为他们的公开内容的范围。已经努力确保关于数字(例如,量、温度等)的准确性,但是应当考虑一些误差和偏差。除非另有指示,否则份数是重量份数,温度是以℃计或者是环境温度,并且压力是大气压或接近大气压。
实施例1.
催化剂制备。通过干法浸渍(dryimpregnation)或初湿含浸浸渍法来制备单金属和双金属ni、fe、co催化剂。ni(no3)2.6h2o(acrosorganics)、fe(no3)2.9h2o(alfa-aesar)和co(no3)2.6h2o(acrosorganics)被用作前体。热解二氧化硅(cab-o-sil-eh-5未处理的sio2;cabot)、氧化铝(其中表面积=100-200m2/g的al2o3γ相;alfaaesar);和沸石(cbv2314或h-zsm-5,其中sio2/al2o3摩尔比率=11.5,并且表面积为约425m2/g;zeolystinternational)被用作载体材料。将金属前体的水溶液(对应于60wt%的金属负载量)浸渍到载体上。将样品在130℃在烘箱中干燥持续过夜(16h)。该合成后的催化剂在马弗炉中经受煅烧并且然后在ar流(70mlmin.-1)中在10%的h2中还原。将单金属ni/sio2在500℃、5℃min.-1煅烧持续10h,并且在450℃、10℃min.-1还原持续4h;将fe/sio2在500℃、5℃min.-1煅烧持续10h,并且在700℃、10℃min.-1还原持续4h;将co/sio2在450℃、5℃min.-1煅烧持续3h,并且在580℃、10℃min.-1还原持续4h。将双金属ni-fe/sio2在500℃、5℃min.-1煅烧持续10h,并且在700℃、10℃min.-1还原持续4h;将ni-co/sio2在750℃、5℃min.-1煅烧持续5h,并且在700℃、10℃min.-1还原持续2h;将fe-co/sio2在450℃、5℃min.-1煅烧持续3h,并且在580℃、10℃min.-1还原持续4h。本工作涉及单金属ni、fe、co催化剂和双金属ni-fe、ni-co和fe-co催化剂。通过共浸渍法以不同的摩尔比(诸如1:1、1:2、2:1、9:1、4:1、1:9)来制备双金属催化剂。催化剂被记为xm-yn,其中,m、n和x、y分别代表金属和摩尔数。
反应器设备。甲烷催化分解在大气压下在固定床流动反应器(10mm内径和44.5cm长的石英管)中进行。在典型的测试中,将0.1g催化剂置于反应器床中,并且用固定在催化剂床处的k型热电偶来测量反应温度。在活性测试之前,催化剂在它们相应的温度经受还原,并且然后用n2吹扫持续30min。然后在n2(70mlmin.-1)中将温度升高至指示的温度,例如典型地约650℃至约750℃(10℃min.-1),并将进料切换至反应物气体(30%ch4/n2,70mlmin.-1),并且这里获得了42000h-1的空速。出口气体的组成通过在线气相色谱仪(perkinelmerarnel,clarus500)来确定,该在线气相色谱仪配备有热导检测器,该热导检测器具有hayesepn60/80、hayesept60/80、分子筛5a45/60和分子筛13x45/60填充柱。gc数据使用totalchromworkstation软件处理。在分析前,gc用标准气体良好地校准。测试催化剂的再现性,并且结果具有±5%的误差。在甲烷分解反应(1h)之后,使用的催化剂在120℃干燥持续2h并且被表征。将所形成的碳纳米管沉积在催化剂上,并且还使用若干种表征技术来表征。
分析方法。使用配备有tcd检测器的micromeriticsautochemhp化学吸附分析仪对催化剂进行程序升温还原(tpr)。将催化剂在热200℃脱气持续1h。在冷却至室温后,将温度在10vol%的h2/ar(50ml·min.-1)中以10℃/min的线性加热速率升高至850℃,并且记录tcd信号。在700℃反应后获得的具有碳沉积的催化剂通过x射线衍射(xrd)来表征。使用cuk辐射在panalyticalx’pertpro上进行xrd测量。步进扫描(stepscan)在10°-90°的范围内进行并且扫描速率为5°min-1。碳材料的形貌和微观结构通过jeoltem-2100透射电子显微镜(tem)来表征。通过在异丙醇中声处理用过的催化剂来制备样品,并且将悬浮液滴到cu-tem网格上用于分析。拉曼实验在环境大气和室温在renishawinvia拉曼光谱仪中进行。使用532nm的绿色激发线来记录光谱。热重分析在5%o2/he气氛中、在从150℃至700℃、在2℃min-1的加热速率使用ta_sdt-650_discovery型仪器进行。
新鲜的和用过的单金属载体的金属催化剂和双金属载体的金属催化剂的性质。使用如本文所描述的xrd和tpr技术来研究催化剂性质。所有金属(ni、fe和co)的特征峰使用xrd分析来识别(图15-图21)。由于对于负载的金属(40%sio2载体上的60%金属)的高强度峰,sio2载体的无定形峰在衍射图中不可见。金属的尖锐峰证实了活性元素(ni/fe/co)的结晶相,并且对应的2θ值表示它们的金属态。在2θ°=44.6°、52.0°、76.6°(jcpdsno.04-850)处观察到ni特征峰;fe特征峰在2θ°=45.1°、65.5°、82.8°(jcpdsno.65-4899)处,并且对于co,峰在2θ°=44.4°、51.6°、76°(jcpdsno.15–0806)处。对于具有不同比率的双金属ni-fe催化剂,数据示出了峰位移,这表示合金形成(图15b)。在ni-fe本体合金中观察到作为fe含量的函数的从fcc相到bcc相的转变(l.-h.zhu等人,j.mater.sci.,2001,36,5571–5574;j.f.valderruten等人,j.phys.condens.matter,2008,20,485204)。对于单金属ni催化剂(111),平面表示fcc相,并且对于单金属fe催化剂(110),平面表示bcc相。在双金属催化剂中,当fe被引入至富ni的体系(9ni-1fe)时,观察到对应于ni-fe合金的fcc相的单组衍射图。还发现该峰朝向更低的2θ值移动,收敛于nifcc相的衍射图。对于4ni-1fe催化剂,fcc合金相(111)更占优势,具有比bcc合金相(110)更高的强度。随着fe含量、2ni-1fe和1ni-1fe的进一步增加,发现fcc合金相移动至较低的2θ值。对于1ni-2fe,ni原子被引入至fe晶格,并移动至较低的2θ值,指示晶格膨胀是ni-fe合金的bcc相中合金形成的结果(n.moghimi等人,j.phys.chem.c,2013,117,4852–4858)。类似地,对于ni-co双金属催化剂,使用xrd分析证实了合金形成(图17)。在2θ°=44.4°处观察到单金属co的fcc相(111),其朝向较高的2θ值(ni相)轻微移动,表示ni-co合金形成。同样对于fe-co金属体系,观察到合金形成,其中fe的bcc相朝向较低的2θ值移动,表示co的fcc相和fe-co合金形成(图19)。除了1fe-2co和1fe-9co,所有其他催化剂仅示出单一合金相。但是对于1fe-2co和1fe-9co,数据示出co的fcc相和fe的bcc相两者。xrd分析可以证实所制备的双金属ni/fe/co催化剂中合金的形成,这从而增加催化剂在反应条件下的稳定性。
在反应后,所使用的催化剂再次通过xrd分析来表征(图16、图18、图20)。大多数催化剂在其金属形式下是稳定的,但是单金属fe催化剂(图16)已经经历氧化,这非常明显,当暴露于空气时,fe易于氧化。但是对于双金属催化剂,观察到合金形成已经有助于防止金属的氧化,并且从而增加其寿命或稳定性。根据xrd数据,使用scherrer方程(表1、表2和表3)计算反应之前和反应之后的金属纳米颗粒的平均微晶尺寸。发现新鲜的ni催化剂的平均微晶尺寸为25nm,而对于新鲜的fe催化剂,平均微晶尺寸为29nm。当制备ni-fe双金属催化剂时,数据示出微晶尺寸的显著减小,对于2ni-1fe催化剂,微晶尺寸甚至减小直到9nm。在新鲜的co催化剂的情况下,发现微晶尺寸为21nm。对于新鲜的ni-co双金属催化剂,发现微晶尺寸在ni催化剂和co催化剂的微晶尺寸之间,为22-25nm。同样对于fe-co催化剂,数据示出17-28nm的微晶尺寸,其在fe和co单金属催化剂的微晶尺寸的范围内。即使在甲烷分解之后,用过的ni-fe/ni-co和fe-co催化剂的微晶尺寸也几乎没有增加。因此,纳米颗粒没有太多团聚,这表明催化剂的稳定性质。奇怪的是,对于单金属fe催化剂,微晶尺寸从29nm减小到13nm,这可以是因为一部分fe位点在暴露于空气时被氧化成铁氧化物。
为了研究合成的催化剂的还原性,进行了h2-tpr实验,并且结果在图21-图23中示出。tpr结果也支持了在双金属催化剂中的合金形成的xrd数据。在单金属ni催化剂中,观察到两个还原峰,一个在367℃左右,并且另一个在470℃(图20)。第一个峰对应于与sio2载体弱相互作用的本体nio的还原,并且第二个弱还原峰是针对与sio2载体具有非常强的相互作用的nio物质的还原(n.wang等人,int.j.hydrogenenergy,2012,37,19–30)。对于单金属fe催化剂,典型的tpr曲线分别在470℃、576℃和727℃左右具有三个峰(图21)。这些对应于三个连续的还原步骤,α-fe2o3→fe3o4→feo→fe(p.ubilla等人,j.chil.chem.soc.,2010,55,35–38;j.yang等人,j.mol.catal.achem.,2006,245,26–36)。在单金属co催化剂中,以306℃为中心的低温还原峰和在360℃处的第二个峰被认为是尖晶石的两步还原,即co3o4→coo→co(图23)。在433℃左右还观察到另外的肩峰(p.jana等人,int.j.hydrogenenergy,2012,37,7034–7041)。然而,高还原温度峰的缺失示出了钴与sio2载体的相互作用不强。双金属ni-fe催化剂的tpr研究示出,随着ni含量的增加,fe的高温还原峰朝向ni物质的还原温度移动(图21)。对于fe-co催化剂,示出了co含量的增加降低了催化剂的还原温度或co有助于fe的还原(图23)。当fe以高浓度存在时,催化剂呈现出fe的性质。因此,观察到co还原温度的升高(d.j.duvenhage和n.j.coville,appl.catal.agen.,1997,153,43–67;r.j.kaleńczuk,catal.letters,1995,33,255–268)。ni-co催化剂的tpr曲线(图22)指示,co3o4比nio更容易还原,而ni含量的增加阻碍了co3o4的还原。因此,co的并入应当提高nio的还原性。
催化剂组合物对甲烷分解和单金属催化剂的影响。使用如本文描述制备的单金属催化剂进行研究。数据示出,尽管ni/sio2示出50%的高ch4转化率,但它开始失活并且在反应的60min内达到40%的转化率(图1)。类似地,测试了单金属fe/sio2,其示出11%的非常低的初始活性,并逐渐地失活至4%的ch4转化率。单金属ni/sio2是非常活跃的催化剂,然而,失活相对迅速地非常快发生。测试了不同载体材料对单金属催化剂的性能的影响。由二氧化硅(sio2)、氧化铝(al2o3)和沸石上的基于fe的催化剂生产氢的效率在图33中示出(60wt%的fe和40wt%的所指示的载体材料)。数据示出,载体材料对氢气产生的效率具有显著影响,其中在fe/al2o3组合物的存在下发现最有效的氢气产生。还确定了基底生长的碳纳米管的效率可以通过载体材料的类型来改变,例如,与所公开的双金属催化剂组合物相比,使用所公开的单金属催化剂组合物最有效地发生基底生长的碳纳米管。在所公开的单金属催化剂组合物中,确定fe/al2o3组合物提供了比fe/sio2组合物更高质量的基底生长的碳纳米管。在图32a-图32b中示出了使用fe/al2o3组合物形成的碳纳米管的代表性图像。
催化剂组合物对甲烷分解和碳纳米管生长的影响—ni-fe双金属催化剂。因此,fe促进剂的添加对用于甲烷分解反应的ni/sio2催化剂活性的影响已经被描述。研究了若干种ni-fe摩尔比(9:1,4:1,2:1,1:1,1:2)。在tos=30min时,4ni-1fe/sio2、2ni-1fe/sio2呈现出与单金属ni/sio2催化剂相似的活性(~50%的ch4转化率),但是后来观察到,即使在tos=60min之后,4ni-1fe/sio2催化剂仍保持其活性。而2ni-1fe/sio2像ni/sio2催化剂一样失活。还用1ni-1fe/sio2和1ni-2fe/sio2进行反应,与上述催化剂相比,示出非常低的ch4转化率(16%-20%),但是它们在整个反应时间内保持活性。因此得出结论,ni-fe双金属催化剂中的高ni含量呈现出较高的转化率,同时有助于提高催化剂的稳定性或寿命。所以,制备了9ni-1fe/sio2催化剂,其示出了60%的优异的转化率,在本文所研究的所有ni催化剂和fe催化剂中最高,并且即使在反应的60min时其活性也非常稳定。发现对于具有较高ni含量的催化剂诸如9ni-1fe/sio2、4ni-1fe/sio2、2ni-1fe/sio2,h2收率在30%-40%的范围内,而其余的催化剂仅示出5%-12%的h2收率(图24)。计算每克催化剂所形成的碳的量,发现在9ni-1fe/sio2、4ni-1fe/sio2、2ni-1fe/sio2上碳的量近似为2.3g-2.5g,并且对于其余的催化剂,碳的量小于0.4g(图25)。
催化剂组合物对甲烷分解和碳纳米管生长的影响—ni-co双金属催化剂。在本节中研究了单金属和双金属ni和co催化剂上的甲烷转化率。在反应条件下,co/sio2示出了37%的ch4转化率,但在反应的5min内失活至9%(图2)。ni/sio2已经示出了良好的初始活性,但在反应过程期间逐渐失活。因此,为了提高co/sio2的活性以增加ni/sio2催化剂的稳定性,本文公开了具有多种ni:co摩尔比的双金属组合ni-co催化剂,诸如sio2载体上的9ni-1co、4ni-1co、2ni-1co、1ni-1co、2ni-1co、1ni-9co。类似于ni-fe催化剂,ni-co组合也示出了55%的最大转化率。ni-co催化剂诸如9ni-1co、4ni-1co、2ni-1co和1ni-1co示出类似的50-55%的初始转化率,并且在整个反应中保持其活性。还测试了具有较高的co含量的催化剂1ni-9co,其示出53%的初始转化率,但是在反应的15分钟内。观察到ni-co双金属催化剂中的高ni含量呈现出更高的转化率,并且co作为促进剂的存在有助于增加催化剂寿命。
发现对于具有较高的ni含量的催化剂,诸如9ni-1co/sio2、4ni-1co/sio2、2ni-1co/sio2,h2收率在38%-40%的范围内。而co/sio2和1ni-9co/sio2仅示出28-33%的初始h2收率,其在反应的5min内降低到6%(图26)。计算每克催化剂所形成的碳的量,发现在9ni-1co/sio2、4ni-1co/sio2、2ni-1fe/sio2上碳的量近似地为2.3g-2.5g,并且对于co/sio2和1ni-9co/sio2催化剂,碳的量为0.15g-0.27g。(图27)。
催化剂组合物对甲烷分解和碳纳米管生长的影响—fe-co双金属催化剂。单金属和双金属fe-co催化剂上的甲烷分解反应在本节中解释(图3)。以多种fe:co摩尔比(诸如9fe-1co、2fe-1co、1fe-1co、1fe-2co、1fe-9co)在sio2载体上制备双金属fe-co/sio2催化剂。观察到1fe-2co/sio2示出51%的最高ch4转化率,但经历了剧烈的失活,并且在反应的60min时示出仅15%的转化率。在本文研究的催化剂中,即使活性不是极好,但稳定性优于9fe-1co/sio2和4fe-1co/sio2。用1fe-2co/sio2观察到29%的最大h2收率(图28,支持信息)和0.8g的碳收率(图29)。
还原的催化剂和氧化的催化剂的活性的比较。通常,过渡金属催化剂(ni/fe/co)以其还原形式(ni0/fe0/co0)用于通过甲烷分解的碳纳米管合成。然而,存在一些文献报道,其中他们已经表明可能没有必要预还原用于反应的催化剂(w.qian等人,appl.catal.agen.,2004,258,121–124)。在本文公开的研究中,已经使用了氧化形式和还原形式的9ni-1fe/sio2催化剂,因为它对于甲烷分解反应示出非常好的活性(图30)。这两种催化剂在反应的初始点都示出相似的活性(约60%)。由氧化的催化剂示出的活性可以是由于晶格氧的原位还原,晶格氧为甲烷分解供应能量,这是吸热过程。除此之外,还可以存在所形成的h2的原位消耗,这有助于平衡朝向甲烷分解步骤或碳形成的移动(w.qian等人,appl.catal.agen.,2004,258,121–124)。如图1中所示,还原的9ni-1fe/sio2催化剂的活性示出恒定的活性直到反应结束,但是氧化形式的催化剂在反应时间的60min内从60%的转化率逐渐失活至41%。对于两种催化剂,每克催化剂所形成的碳的量也在2.2g-2.5g的范围内。因此,总的来说,在我们的情况下,即使氧化形式的催化剂催化甲烷分解成碳和h2,但是与它们的还原形式相比,催化剂的稳定性较低。这可能是因为还原形式由于它们的合金相而稳定,这有助于增加它们的寿命和稳定性。
碳纳米管的性质—xrd分析。所使用的催化剂的所有xrd图均通过在2θ=26.2°处的非常强烈的峰(002)示出了石墨碳的存在(图15、图17和图19)。还识别了所有的特征金属和合金峰,不存在在这些催化剂中的任何金属碳化物形成的明显证据。根据xrd图,很难区分碳纳米管和类似的石墨结构之间的微观结构特征,因为它们的特征峰是重叠的(w.z.zhu等人,mater.chem.phys.,2003,82,638–647)。此外,使用用于石墨峰的布拉格方程(d=λ/2sinθ)计算碳纳米管的d间距,发现该d间距为0.34nm并且其与两个石墨层的距离(0.3354nm)良好相关,这意味着在所有催化剂上生长的碳的高结晶度。(002)衍射峰的强度与石墨化的程度有关(图4)。因此,较低的强度表示较低石墨化的材料(c.-m.chen等人,carbonn.y.,2006,44,1808-1820)。在ni-fe双金属催化剂的情况下,数据示出9ni-1fe/sio2或具有最高ni含量的双金属催化剂产生具有最高石墨化程度的碳纳米管,并且其石墨化程度随着催化剂中fe的量的增加而降低。
ni-fe催化剂的石墨化程度依次为9ni-1fe>ni>4ni-1fe>2ni-1fe>1ni-2fe>fe。对于ni-co双金属催化剂,4ni-1co/sio2产生具有最高石墨化程度的碳纳米管,并且石墨化程度依次为4ni-1co>2ni-1co>ni>1ni-1co=9ni-1co>1ni-9co>co。对于fe-co催化剂,与基于ni的催化剂相比,碳纳米管峰的整体强度非常低,并且整组fe-co催化剂产生较低石墨化的碳纳米管。因此,通常双金属催化剂中的ni含量影响在甲烷分解反应期间形成的碳的石墨化程度。
碳纳米管的性质—热重分析(tga)。碳纳米管的热稳定性使用热重分析法来研究。研究了所使用的催化剂诸如9ni-1fe/sio2、9ni-1co/sio2和1fe-2co/sio2,因为这些催化剂示出比其余催化剂更好的活性(图5)。这些催化剂在2%o2/he气氛中被分析。沉积在催化剂、无定形碳纳米管或碳纳米管上的碳在o2气氛中分解成co或co2。在某些情况下,存在催化剂物质还在这些条件下被氧化的可能性。观察到在200℃-350℃的温度范围内没有热降解,这对应于无定形碳。因此,证实了,沉积在这些催化剂上的碳本质上不是无定形的。在所有这些催化剂中观察到的重量损失对应于沉积在催化剂上的碳纳米管。从碳纳米管的tga分析可以理解,碳的降解温度越高,其稳定性越高。对于9ni-1fe/sio2,热降解开始于500℃左右,并且存在75%的重量损失。对于9ni-1co/sio2,降解从450℃开始,其中重量损失为70%。对于1fe-2co/sio2催化剂,在350℃左右的温度,存在重量2%的略微增加,这可能是由于催化剂中存在的fe的氧化。在410℃左右,观察到45-50%的重量损失,这低于9ni-1fe/sio2催化剂和9ni-co/sio2催化剂。9ni-1fe/sio2、9ni-co/sio2和1fe-2co/sio2的起始温度分别为500-660℃、450-650℃和410-640℃,这指示在9ni-1fe/sio2催化剂上形成的碳纳米管具有较高的石墨化程度以及较少的碳纳米管上的缺陷。因此,可以得出结论,起始温度和终止温度的较小差异指示了高度石墨化的碳纳米管的形成。还应当理解,在这些催化剂上的甲烷分解期间不存在无定形碳形成,并且与9ni-1fe/sio2催化剂和9ni-co/sio2催化剂相比,在1fe-2co/sio2上形成的碳的量最小。
碳纳米管的性质—拉曼分析。进行拉曼光谱研究以了解碳的质量和结晶度(图6、图7和图8)。对于所有催化剂观察到两个有区别的带,在1336cm-1处的d带和在1570cm-1处的g带。d带表示无序的碳或无定形碳,并且g带表示结晶碳(q.weizhong等人,appl.catal.agen.,2004,260,223–228)。从tga分析证实,没有无定形碳沉积在催化剂上(图5)。因此,强度比id/ig解释了碳纳米管的石墨化程度和结晶度(a.e.awadallah等人,appl.surf.sci.,2014,296,100–107)。催化剂具有带有最高结晶度的最低id/ig碳。对于ni-fe催化剂(图6),数据示出对于9ni-fe、4ni-1fe、ni、1ni-2fe,id/ig值最低,示出相似的id/ig值(0.829-0.874),结晶度比具有较高的id/ig值(0.944-1.26)的2ni-fe、1ni-1fe和fe催化剂更好。
ni-co催化剂的拉曼光谱(图7)表明,9ni-1co给出了具有最低的id/ig(0.765)的大多数结晶碳纳米管,而ni、4ni-co、1ni-1co示出了类似的id/ig(0.868-0.883)。数据没有示出co和1ni-9co催化剂的任何有区别的带,因为对于这些催化剂形成的碳纳米管的量低于检测限值。对于4fe-1co,计算的结晶度最高(id/ig=0.896),而fe、9fe-1co、2fe-1co示出类似的id/ig(0.983-0.99)。由于所形成的碳纳米管的低结晶度,剩余的催化剂比率诸如1fe-1co、fe-2co、fe-co催化剂4fe-1co形成具有最高的id/ig(1.019-1.042)的碳纳米管。
碳纳米管的性质—tem分析。使用tem技术研究了碳纳米管的结构形态、粒径、直径和生长。该分析限于来自每组双金属催化剂(ni-fe/ni-co和fe-co)的某些催化剂,这些催化剂在甲烷分解反应中示出最佳的性能和稳定的性质(图9)。因此,对所使用的9ni-1fe/sio2、9ni-1co/sio2和1fe-2co/sio2催化剂进行tem分析。观察到,在我们的实验中形成的碳纳米管被发现是mw碳纳米管,这是由于活性位点金属纳米颗粒的大微晶尺寸,其通过xrd技术来证实。在所有这些催化剂上形成的碳纳米管示出非常拥挤的密集的聚集和缠结的纤维。由于生长的空间竞争,碳纳米管以随机的方向生长(g.wang等人,energy&fuels,2013,27,4448–4456)。碳纳米管的长度取决于工艺的持续时间,因此为了获得更长的丝,必须延长反应的持续时间。
对于9ni-1fe/sio2催化剂,观察到大部分所形成的碳纳米管具有100-120nm。管的壁非常厚,其中石墨层非常紧密地堆叠。令人惊讶的是,ni-fe催化剂示出碳纳米管的“尖端生长”,而碳纳米管生长,它带着金属纳米颗粒一起并且位于碳纳米管的尖端处。金属纳米颗粒的形状是“锥形的或梨形的”,朝向端部逐渐变细,并与管状轴线形成角度。从hr-tem观察到,所形成的碳纳米管的壁为“鱼骨状或人字形”结构,其中石墨烯层相对于纤维轴线倾斜地堆叠。在这些类型的碳纳米管中,石墨平面以相对于纳米管的轴线成一定角度形成,并且因此在这些碳纳米管中存在边缘平面位点/缺陷的更高的可能性(c.e.banks和r.g.compton,analyst,2006,131,15–21)。对于9ni-1co催化剂,形成了具有50-60nm直径的mw碳纳米管,并且在尖端处还具有金属纳米颗粒。但是从hr-tem观察到,这些碳纳米管的壁具有平行的形态(石墨平面平行于管状轴线布置)。但是在这些催化剂中形成的某些纤维在尖端处没有金属纳米颗粒(图31),碳纳米管在其基底处用金属生长(基底生长)。所以,9ni-1co/sio2产生了尖端生长的碳纳米管和基底生长的碳纳米管的混合物。
在fe-co催化剂的情况下,mw碳纳米管形成有100-125nm的直径,还示出了尖端生长的碳纳米管和基底生长的碳纳米管的混合物(图9和图31)。hr-tem已经证实用于这些催化剂的碳纳米管壁的平行形态。
基底生长的碳纳米管的合成。来自催化剂筛选研究的数据示出,使用单金属和双金属ni/fe/co催化剂,60%的fe/sio2组合物在甲烷分解期间提供了选择性地形成基底生长的碳纳米管。因此,fe/sio2催化剂被用于接下来的系列研究中。本文公开的数据示出,fe/sio2具有非常低的甲烷转化率,为11%,并且最终在t=650℃、tos=60分钟、ghsv=42000h-1时降低至4%。因此,反应条件需要创造性的改进,以便获得h2和碳纳米管的更好的转化率和收率。
在多个温度t=650℃、700℃、750℃和800℃,在fe/sio2催化剂上研究了温度对碳纳米管的基底生长的影响(图10)。观察到,在t=750℃时,甲烷转化率为50%,比在t=700℃时的47%好。但是在15分钟的反应后,甲烷转化率在两种情况下是相似的。因此,对于未来的研究,优选t=700℃。使用tem分析证实了基底生长的碳纳米管的形成。观察到,金属纳米颗粒保留在载体上,而碳纳米管从基底生长(图11)。
因此,60%的fe/sio2催化剂被证明能够通过甲烷分解合成基底生长的碳纳米管。为了证实fe/sio2催化剂的这种性质,如本文所公开的进行催化剂再生研究。在t=650℃、tos=60min、ghsv=42000h-1的第一次甲烷分解循环之后,将所使用的催化剂在500℃使用10%的o2再生持续30min。沉积在催化剂上的碳以co2的形式烧掉,这使用gc分析被证实。实验的第二个循环在相同的反应条件下进行,并且发现活性在两个循环中相同(图12)。在第二个循环中形成的碳纳米管使用tem来表征,这证实即使在第二个循环中也形成碳纳米管的基底生长(图13)。这可能是因为在反应的过程期间,fe纳米颗粒的颗粒的活性位点没有被聚集。进行碳纳米管的第一个循环和第二个循环两者的拉曼分析,这示出了在第二个循环中,对应于无序的碳纳米管的d带的强度高于对应于碳纳米管的结晶度的g带(图14)。因此,即使在再生研究之后,fe/sio2的活性没有降低,然而,发现该催化剂对碳纳米管形成的选择性在再生之后降低。
该项目的中心主题是甲烷(页岩气的主要组分)到无cox的h2和有价值的碳(诸如碳纳米管)的催化分解。然而,为了实现该目标,必须对反应条件、催化剂性质和由该工艺生成的碳进行彻底的研究。
总之,单金属的和双金属的基于ni/fe/co的催化剂的催化活性、选择性和稳定性被证明用于甲烷分解研究。本文公开了过渡金属催化剂(ni/fe/co),其通过甲烷分解提供尖端生长的碳纳米管和基底生长的碳纳米管两者的合成。本文公开了通过在sio2载体上干法浸渍而制备的新型合成的单金属和双金属ni/fe/co催化剂。xrd和h2-tpr分析证实了在双金属催化剂中合金的形成。在多种摩尔比的基于ni、fe和co的催化剂上,研究了催化剂组合物对甲烷分解和碳纳米管生长的影响。ni-fe双金属催化剂中的高ni含量呈现出较高的甲烷转化率,并且还有助于催化剂的稳定性的增加。在ni-co双金属催化剂的情况下,较高的ni含量还呈现出较高的转化率,并且催化剂中存在的co有助于增加催化剂的寿命。在fe-co催化剂中,对甲烷转化的活性相对地低于ni-fe催化剂和ni-co催化剂。使用xrd、tga、拉曼和tem技术分析了在这些催化剂上形成的碳纳米管的品质。从xrd分析可知,双金属催化剂中的ni含量控制所形成的碳的石墨化程度。从tga分析得出结论,在这些催化剂上的甲烷分解期间不存在无定形碳形成。使用拉曼分析来计算碳的品质,即结晶度(degreeofcrystallinity)。tem分析有助于了解碳纳米管的不同形态、直径、壁类型(平行类型或鱼骨类型)以及还有生长机制,诸如在不同催化剂上的尖端生长和基底生长。应当理解,ni-fe催化剂选择性地产生具有鱼骨状壁图案的尖端生长的碳纳米管,而ni-co催化剂和fe-co催化剂形成具有平行壁图案的尖端生长的碳纳米管和基底生长的碳纳米管的混合物。
对甲烷分解的先前的催化研究主要集中在这些金属上,这些金属单独地作为用于碳纳米管形成和氢气产生的催化剂。此外,缺乏关于金属的类型和在甲烷分解期间形成的碳纳米管的性质之间的关系的信息。在用于甲烷分解的相同反应条件下,使用多种ni/fe/co催化剂的组合,我们的工作确实显著地更加普遍。本文还公开了在fe/sio2上合成选择性地基底生长的碳纳米管,这种碳纳米管被认为易于收获而不牺牲催化剂位点。所公开的组合物和方法通过甲烷的催化分解提供了碳纳米管和无cox的h2的新型合成。
实施例2.
催化剂制备。通过干法浸渍或初湿含浸浸渍法来制备基于fe的催化剂。fe(no3)2·9h2o(alfaaesar)被用作金属前体。γ(γ)al2o3(alfaaesar)、热解sio2(cab-o-sil-eh-5未处理的sio2,cabot)和h-zsm-5沸石(cbv-5524g,sio2/al2o3摩尔比率=50,zeolystinternational)被用作催化剂载体。将金属前体的水溶液(60wt%的金属负载量)浸渍在载体(40wt%)上,并且在130℃在干燥烘箱中干燥持续过夜(16h)。此外,将合成后的样品在500℃煅烧持续10h,并且然后在ar流(70mlmin-1)中在10%的h2中还原。所获得的催化剂被命名为fe/al2o3、fe/sio2和fe/h-zsm-5。除了60wt%的fe/al2o3之外,还制备了10wt%和30wt%的fe/al2o3。这些催化剂被表示为xfe/载体(x意指fe的重量%负载量)。
反应器设备。甲烷催化分解反应在大气压(1巴)在固定床流动反应器(具有10mm内径的44.5cm长石英管)中进行。在置于反应器床中的0.1g催化剂的情况下进行实验,并且用n2吹扫持续30min。在活性测试之前,将煅烧的催化剂在10%的h2/n2流(70ml/min)中在700℃(10℃/min)还原持续4h。催化剂用n2吹扫持续30min,并且在n2(70ml/min)中将温度升高至700℃(10℃/min)。此外,将进料切换到具有42000h-1的空速的反应物气体(30%ch4/n2,70ml/min)。出口气体的组成通过在线气相色谱仪(perkinelmerarnel,clarus500)来分析,所述在线气相色谱仪配备有热导检测器和一系列填充柱(hayesepn60/80、hayesept60/80、分子筛5a45/60和分子筛13子45/60)。gc数据使用totalchromworkstation软件处理。在分析之前,gc用标准气体良好地校准。测试催化剂的再现性,并且结果具有±5%的误差。在催化剂上形成的cnt通过对其称重被量化。
表征技术。催化剂样品通过若干种仪器来表征。在700℃反应后获得的具有碳沉积的催化剂通过x射线衍射(xrd)来表征。使用cuk辐射在panalyticalx’pertpro上进行xrd测量。碳纳米管的拉曼光谱是通过renishawinvia拉曼显微镜在5mw激光功率下用532nm的激发波长使用拉曼光谱进行的,并且对于每个光谱累积三次扫描。扫描电子显微镜(sem)图像通过配备有oxfordincaeds的jeoljsm-7600fsem来观察。透射电子显微镜(tem)的研究采取了jeoljem-2100tem并且操作电压是200kv。用过的催化剂和纯化的催化剂的热重分析(tga)在sdt650仪器(tainstruments)中进行。样品在5%的o2/n2中以5℃/min的恒定加热速率加热直到900℃。同时,记录重量损失和氧化温度。x射线光电子能谱(xps)。
载体的效果。研究了fe催化剂在多种载体(fe/al2o3、fe/h-zsm-5和fe/sio2)上对于在700℃甲烷分解的催化活性。来自甲烷分解的主要产物是氢气和碳cnt。使用gc分析进行h2的定量,同时在催化剂上沉积的碳基于在反应后催化剂的重量增加来计算,并且使用不同的技术来表征。cnt在两种载体上的生长速率表明,催化剂形态可以极大地影响催化剂的活性,因为在ccvd中,速率确定步骤是催化剂颗粒对烃的吸附和分解。在该研究中,观察到ch4的转化率取决于载体的类型,诸如al2o3、sio2和h-zsm-5。对于fe/zsm-5和fe/sio2,初始转化率分别为50%和36%(参见图35)。即使fe/al2o3具有较高的初始转化率(58%),在反应的1h内活性降低(20%)。在fe/h-zsm-5催化剂上,初始转化率也很高(50%),但存在其活性在15min内降低到15%,但在整个反应时间内保持该转化率。类似地,具有36%的初始转化率的fe/sio2降低到9%。每种催化剂的氢气收率与甲烷转化率相似,其遵循相似的趋势。fe/al2o3指示约26%的氢的最高初始收率,并且对于fe/zsm-5和fe/sio2,氢的最高初始收率分别为24%和20%,如图35中所示。计算每克催化剂所形成的碳的量,发现对于fe/al2o3催化剂,其近似为0.785g/g催化剂,而对于fe/zsm-5和fe/sio2,其仅为0.1g/g催化剂。
fe负载量对al2o3载体的影响。研究了在多种fe负载量下,随着运行时间,用于fe/al2o3的甲烷转化的催化活性。fe负载量为10wt%、30wt%和60wt%,其与分别为18%、40%和58%的初始转化率相关联。转化率随着fe的负载量的增加而增加,但在15min后,所有催化剂开始逐渐失活。30%fe/al2o3催化剂的转化率从40%下降到18%,并且10%fe/al2o3的转化率下降到10%。此外,60%fe/sio2和30%fe/al2o3具有相似的甲烷转化率,并且因此载体明显地影响催化剂的活性。此外,如图36中所示,fe、30%fe/al2o3和10%fe/al2o3的不同负载量分别为16%和2%。此外,在其他研究中,确定30%fe/al2o3和10%fe/al2o3的碳收率为约0.38g/g催化剂和0.07g/g催化剂。
使用无机酸的cnt的酸回流分离和纯化。在已经报告的多种纯化方法中,用多种无机酸回流是最常用的处理。35,45,46对于我们的实验,我们已经收集了在根据我们先前的研究的用于甲烷分解的ni-fe双金属(60%9ni:1fe/sio2)催化剂上形成的cnt。21根据我们的实验,由于催化剂的优异性能和非常高的碳收率,选择来自该催化剂的这种cnt用于纯化研究。此外,在我们目前的研究中,我们已经在本文使用的fe/al2o3催化剂上使用了cnt。沉积在催化剂上的cnt的酸回流将除去金属颗粒和碳杂质以及一些化学表面改性。若干因素诸如回流温度、时间和酸的强度决定纯化工艺的有效性(参见:h.hu等人,phys.chem.b107,13838(2003);和y.wang等人,chem.phys.lett.432,205(2006))。在强酸的情况下在较高的温度较长的回流时间导致金属颗粒的完全去除,并且酸还侵蚀cnt的缺陷位点。但是在低的温度和酸浓度的情况下较短的回流时间段导致催化剂颗粒从cnt中的不完全去除。
在实施例2中,采用以下程序用于分离和纯化cnt:在120℃,在连续磁力搅拌下,将0.3g重量的用过的催化剂(9ni:1fe/sio2或fe/sio2)在45ml的3mhno3溶液中回流持续24h和48h。将随后获得的残余物过滤,并用去离子水洗涤,直至ph为中性。据信,回流后留下的酸溶液可能含有溶解的金属颗粒和用过的催化剂上的载体。然后将留下的固体碳(cnt)在120℃干燥过夜(16h),并且使用多种物理化学技术来表征以分析其纯度。
分离的cnt的xrd和拉曼分析。在酸回流后从9ni:1fe/sio2和fe/sio2催化剂中分离的碳使用xrd和拉曼技术来分析。图37a表示在回流24h和48h后,用过的9ni-1fe/sio2与其分离的cnt的xrd图的比较。对于fe/sio2催化剂也是类似的。在这两种情况下,可以观察到在酸回流之前,金属特征峰(ni或fe)以及石墨碳在用过的催化剂的xrd图中清晰可见。
这些金属峰的强度在酸回流的24h后降低,并在回流48h后被完全去除。因此,在48h样品中,我们只能检测到在2θ=26.2和42.9处的石墨碳特征峰,这证实了在我们采用的纯化工艺后分离的碳的纯度。
在图37b中示出了在回流24h和48h后用过的9ni-1fe/sio2及其分离的cnt的拉曼分析。用过的fe/al2o3的id/ig比率为0.755,并且纯化的fe/al2o3的id/ig比率为0.758(24h)和0.73(48h)。用过的9nife/sio2的id/ig比率为0.822,并且纯化的9nife/sio2的id/ig比率为0.834(24h)和0.804(48h)。这些结果示出在纯化之前和纯化之后对于碳的id/ig值没有变化,这指示用于纯化的hno3处理不影响分离的cnt的结晶结构。两个结果都表明,酸处理持续48h有效地从cnt中去除了用过的催化剂的金属和载体残余物,并且保持了cnt的结晶性质;在纯化后获得。
tem分析。图37c示出了在甲烷催化分解后形成的cnt的代表性tem图像。观察到来自催化剂(9ni-fe/sio2)的玉米形金属纳米颗粒被包封在所形成的cnt的尖端处。在使用酸回流纯化后,从cnt中除去金属纳米颗粒,在纳米管的尖端处留下开口(图37d)。因此,tem分析证实了在酸回流后金属纳米颗粒的去除。
实施例3.
材料。分析级化学品,包括alcl3·6h2o、ni(no3)2·6h2o、co(no3)2·6h2o、乙醇(无水)和环氧丙烷(po),购自acrosorganics,并且按所接收到的使用,而无需进一步纯化。用于制备金属溶液的去离子水。
ni/al2o3和co/al2o3气凝胶催化剂的合成。ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂通过溶胶-凝胶法来合成。本节描述了作为特定实例的2g50wt%的ni/al2o3气凝胶催化剂(50wt%的金属在50wt%的al2o3载体上)的制备。在制备中,将4.74g的alcl3·6h2o与4.95g的ni(no3)2·6h2o混合,随后在剧烈搅拌下添加10ml的去离子水和15.5ml的无水乙醇持续30min。将含有溶液的烧杯密封,并且然后在80℃在油浴中加热持续1h。将溶液冷却至室温,并转移至冰浴,向冰浴中添加16ml的环氧丙烷(购自acrosorganics)。溶液在没有搅拌的情况下在室温放置持续35分钟,以允许形成气凝胶。此后,湿凝胶用去离子水洗涤三次,并在无水乙醇中浸泡过夜,以允许乙醇进入孔中。将湿凝胶真空干燥持续9h,随后在600℃煅烧持续8h。在甲烷催化分解测试之前,ni/al2o3气凝胶催化剂在450℃还原持续2.5h。对于co/al2o3气凝胶催化剂,制备方法基本上与ni/al2o3气凝胶催化剂中使用的相同,除了co/al2o3气凝胶催化剂在500℃煅烧持续3h并在600℃还原持续2.5h。
催化剂表征。在反应后,所使用的催化剂通过一系列分析仪器来表征。使用cukα辐射在panalyticalx’pertprox-raydiffractometer上进行x射线衍射(xrd)测量,其中步进扫描在10-80扫的范围内进行。碳材料的形貌和微观结构通过透射电子显微镜(tem)在jeoljem-2100上表征。通过在异丙醇和铜网格中声处理用过的催化剂而制备的tem样品用于装载悬浮液。在renishawinvia拉曼光谱仪上进行拉曼分析,其中使用532nm的绿色激发线来记录拉曼光谱。热重分析(tga)在5%的o2/he气氛中使用ta_sdt-650_discovery型仪器进行。将温度以10℃/min的加热速率从150-900℃逐步升高(ramp)。bet分析使用氮气在77k在asap2020仪器上进行。sem图像在hitachis-4700扫描电子显微镜上获得。xps分析在physicalelectronicsphi5000versaprobex-rayphotoelectronspectroscopy上进行。
反应器设备。甲烷分解在固定床反应器中进行。反应器由具有10mm的内径的石英管制备。在每个测试中,将100mg的ni/al2o3气凝胶催化剂或co/al2o3气凝胶催化剂置于反应器中。在测试之前,催化剂如上文所描述的通过氢气还原。用n2气体吹扫后,将反应器温度升高至650℃,然后切换至反应物气体(30%ch4/n2,70ml/min)。甲烷分解反应在42000h-1的ghsv进行。出口气体的组成通过配备有热导检测器的在线气相色谱仪(perkinelmerarnel,clarus500)分析。
不同制备方法对用于甲烷分解的催化剂性能的影响。
ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂的合成及性能测试。图38a和图38b示意性地图示了示例性公开的气凝胶催化剂的合成中涉及的主要步骤。在气凝胶催化剂制备的过程中,选择alcl3·6h2o作为氧化铝的前体,并且乙醇和去离子水被用作溶剂。环氧丙烷(购自acrosorganics)被用作胶凝剂,其消耗来自水合金属物质的质子以促进溶胶-凝胶聚合反应(例如,参见baumanntf等人,chemmater2005;17:395-401;和gashae等人,chemmater2001;13:999-1007)。如图39a中所示出的,在最初的20min反应时间内,60wt%的ni/al2o3气凝胶催化剂呈现出79.2%的最高ch4转化率,但是随着反应的进行,转化率以比50wt%和70wt%的ni/al2o3气凝胶催化剂更快的速率下降。似乎所有三种ni/al2o3气凝胶催化剂在反应的早期阶段期间具有诱导期。在诱导期之后,50wt%的ni/al2o3催化剂示出最高的甲烷转化率。计算每克气凝胶催化剂所形成的碳的量,分别发现在50wt%的ni/al2o3气凝胶催化剂上,碳的量近似为0.63-0.83g,对于60wt%的ni/al2o3气凝胶催化剂,碳的量为3.31-3.39g,并且对于70wt%的ni/al2o3气凝胶催化剂,碳的量为0.53-0.65g。图39b示出了对于所指示的三种ni/al2o3气凝胶催化剂的氢气收率。与如图39a中所示出的运行时间甲烷转化率相一致,氢气收率在最初的20min期间示出快速增加的趋势,然后,随后开始稳定。
在三种不同金属负载量的co/al2o3气凝胶催化剂上进行甲烷分解。如图39c中所示出的,与ni/al2o3气凝胶催化剂不同,在co/al2o3气凝胶催化剂上没有观察到诱导期。三种催化剂的初始活性似乎略有不同,其中60wt%的co/al2o3气凝胶是最高的。在初始20min期间,对于所有三种催化剂,甲烷转化率显著下降,之后其在10%的甲烷转化率水平趋于平稳。在1小时的测试中,与ni/al2o3相比,对于co/al2o3催化剂,甲烷转化率的损失是显著的,这可以通过以下部分中ni/al2o3气凝胶的增强的孔径分布和扭曲度来解释。制氢的运行时间收率与甲烷转化率一致。ni/al2o3气凝胶催化剂呈现出较高的甲烷转化率和碳生产率。如图39d中所示出的,对于co/al2o3气凝胶催化剂,当金属含量为50wt%或60wt%时,h2收率相对高。然而,当气凝胶催化剂中的金属含量达到70wt%时,h2产量将可能由于金属烧结而导致降低。
通过不同方法制备的催化剂的催化性能比较。将ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂在甲烷分解反应中的催化活性与先前报告的通过常规初湿含浸技术合成的催化剂比较(参见ayillath-kutterid等人,catalsci&technol2018)。如图40a和图40b中所示出的,ni/al2o3气凝胶催化剂的催化活性显著高于通过常规初湿含浸技术合成的在al2o3或sio2上的ni、fe、单金属或双金属的催化活性。如图40c中所示出的,co/al2o3气凝胶催化剂的催化活性也高于通过初湿含浸技术合成的其他co或基于co的催化剂的催化活性。如图40d和图40e中所示出的,对于三种ni/al2o3气凝胶催化剂,h2收率测量为在30%-45%的范围内,而其余的其他催化剂仅示出5%-30%的h2收率。如图40f中所示出的,co/al2o3气凝胶催化剂的氢气收率也高于其他co或基于co的催化剂的氢气收率。所有的以上结果表明金属和载体之间的相互作用以及气凝胶催化剂的合成方法可以影响甲烷分解中的催化活性。通过初湿含浸合成的催化剂,不希望受特定理论束缚,被认为经由之间的弱范德华力在金属和载体之间具有主要结合。结合能可能不足够强以将金属保留在表面上,可能发生金属颗粒从载体表面浸出以形成尖端生长的cnt。本文的数据表明催化剂的制备方法以及与制备方法相关的结构可以影响催化剂的性能。
ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂的表征。为了研究ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂的性质,进行了一系列表征分析,包括xrd、tga、拉曼、xps、bet和sem。通过tem来测量cnt的性质,以评估与生长模式和机理的相关性。新鲜的和用过的ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂的xrd图在图41a和图41b中示出。其中的xrd光谱数据示出了与ni、co和al2o3载体相关联的特征峰。一些与al2o3载体相关的峰在xrd光谱中不可见,可能是由于高强度金属峰的存在。金属的尖锐峰指示了活性元素(ni和co)的结晶相的形成。在2θ=37.3°、46.7°、52.8°、66.6°和76.6°(jcpdsno.04-850)处识别出ni特征峰,而在2θ=44.8°、48.2°和77.6°(jcpdsno.15-0806;参见monzóna,等人.cataltoday2006;116:264-70;和lij,zhoul,zhuq和lih.indengchemres2013;52:6647-54)处观察到co特征峰。在反应后,所使用的催化剂再次通过xrd分析来表征。如图41a和图41b中所示出的,与新鲜的催化剂相比,用过的ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂的xrd图通过在2θ=26.2°处的非常强的峰(002)示出了石墨碳的存在(wangg等人,energy&fuels2013;27:4448-56)。由于xrd分析的限制,cnt和类似石墨结构之间的微结构特征难以区分,因为它们的特征峰是重叠的。使用用于石墨峰的布拉格方程(d=λ/2sinθ)计算cnt的d间距。计算出d间距为0.34nm,这与两个石墨层的距离(0.335nm)良好相关,这意味着在所有气凝胶催化剂上形成的碳的高结晶度。(002)衍射峰的强度与石墨化的程度有关。较低的强度指示较少石墨化的材料的存在。在co/al2o3气凝胶催化剂的情况下,与ni/al2o3气凝胶催化剂相比,cnt峰的总强度显得小得多。co/al2o3气凝胶催化剂产生较少石墨化的cnt。
进行拉曼光谱测量以研究碳产品的品质和结晶度。如图42a和图42b中所示出的,观察到两个有区别的带,在1342cm-1处的d带(可能表示无序的碳或无定形碳)和在1575cm-1处的g带(可能表示结晶碳;参见weiq等人,applcatalagen2004;260:223-28)。cnt的石墨化程度和结晶度可以通过id/ig的强度比来测量。具有最低id/ig碳的气凝胶催化剂表示最高的结晶度。60wt%的ni/al2o3气凝胶催化剂的id/ig比略高于50wt%和70wt%的ni/al2o3气凝胶催化剂。70wt%和60wt%的co/al2o3气凝胶催化剂的id/ig值相似。拉曼光谱示出,70wt%的ni/al2o3气凝胶催化剂和70wt%的co/al2o3气凝胶催化剂两者都形成具有最低id/ig(0.835和0.725)的大多数结晶cnt。尽管60wt%的ni/al2o3气凝胶催化剂示出比co/al2o3气凝胶催化剂更高的生产率,但是id/ig并不是更低的,这意味着缺陷cnt的形成(拉曼中的d带)。不受特定理论的束缚,可能的是,ni/al2o3气凝胶催化剂上的cnt的长度比co/al2o3气凝胶催化剂上的cnt的长度长得多。不良的电子转移可能影响结晶并导致cnt的缺陷。本文的数据与下面本文讨论的tem分析一致。
所使用的ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂通过tga来表征,以研究cnt的热稳定性。tga测量在2%的o2/he气氛中进行。如图42c中所示出的,对于ni/al2o3气凝胶催化剂,在150-550℃的温度范围内没有发生热降解,并且对于co/al2o3气凝胶催化剂,在150-400℃的温度范围内没有发生热降解,这可能对应于无定形碳分解。沉积在气凝胶催化剂上的碳似乎不是无定形碳的特征。tga分析表明用过的催化剂上的碳具有结晶结构。如图42c中所示出的,对于50wt%的ni/al2o3气凝胶催化剂,热降解在500℃左右开始,其中重量损失为84%。对于60wt%的ni/al2o3气凝胶催化剂,降解在500℃开始,其中重量损失为87%。类似于60wt%的ni/al2o3气凝胶催化剂,70wt%的ni/al2o3气凝胶催化剂示出78%的重量损失。
发现co/al2o3气凝胶催化剂的tga曲线不同于ni/al2o3气凝胶催化剂。co/al2o3气凝胶催化剂的热降解温度远低于在ni/al2o3气凝胶催化剂上出现的。此外,发现在三种co/al2o3气凝胶催化剂上形成的碳的量远少于ni/al2o3气凝胶催化剂的碳的量,这与催化活性结果的结果相一致。进行xps测量以分析表面元素,包括碳、氧、镍和钴。图42d中示出的结果总体上与文献报告的c1s、o1s、al2p、ni2p3和co2p3光谱的峰的分配相一致(例如,参见monzón等人,cataltoday2006;116:264-70;和lij,papadopoulosc,xujm和moskovitsm.applphyslett1999;75:367-9)。具体而言,ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂的c1s的峰可以分别在284.6ev和288.0ev处解卷积为峰。对于ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂,o1s的核心级xps光谱分别在531.7ev和532.7ev处示出了主峰。在al2p的核心级xps光谱中观察到明显的信号峰,该峰在80ev处。ni2p3的xps光谱在531.7ev和532.7ev处示出两个主峰[23]。对于co2p3,在785.6ev和800.2ev处的两个主峰在co/al2o3气凝胶催化剂的核心级xps光谱中被证实。结果指示在ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂中ni2+离子和co2+离子与al2o3载体的配位。不希望受特定理论的束缚,可能的是,配位形成强的相互作用,这将有助于提高催化活性。
进行sem和bet测量以研究ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂的微观结构、孔径分布和吸附-脱附性质。测量限于60wt%的ni/al2o3和co/al2o3气凝胶催化剂,其在甲烷分解反应中示出最佳性能。如图43a中所示出的,60wt%的ni/al2o3和co/al2o3气凝胶催化剂的等温线非常相似,并且两种气凝胶催化剂的bet表面积分别被测量为324m2·g-1、271m2·g-1。该bet表面积高于其他报告的结晶金属氧化物气凝胶的bet表面积(baumanntf等人,chemmater2005;17:395-401;gaponikn等人,advmater2008;20:4257-62;sanchez-paradinass等人,advmater2015;27:6152-6;和sorensenl,strousegf,以及stiegmanae.advmater2006;18:1965-7)。
如图43b中所示出的,60wt%的ni/al2o3气凝胶催化剂的barrett-joyner-halenda(bjh)孔径被定位在~10-25nm的区域中,而对于60wt%的co/al2o3气凝胶催化剂,孔直径主要分布在~5-15nm处。尽管退火减小了表面积,但复合气凝胶催化剂在650℃热处理后仍呈现出高的表面积,并且在介孔范围呈现出宽的孔径分布。我们已经知道,介孔催化剂示出比微孔催化剂的活性更高的活性。纳米复合材料介孔ni/al2o3和co/al2o3气凝胶催化剂具有较高的表面积和良好的吸附性质,这有助于提高催化活性和氧化再生。相比之下,通过常规的初湿含浸技术合成的催化剂在制备过程期间不能保持多孔结构。因此,结果指示,通过不同方法合成的催化剂对其性质具有很大影响。sem图像示出,对于60wt%的ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂,在胶体颗粒之间形成具有从几十纳米到几百纳米的间隙距离的通道。在cnt和催化剂颗粒之间形成交联的cnt。如图43c和图43d中所示出的,sem图像指示cnt是交织的,并且与载体颗粒组合以形成网状结构,并且这种交联的结构是非常多孔的。似乎催化剂的曲折度对cnt的生长非常重要。
催化剂的不同制备方法对cnt的生长模式的影响。如图44a的部分中所示出的,对于60wt%的ni/al2o3气凝胶催化剂和70wt%的ni/al2o3气凝胶催化剂,发现所形成的cnt是多壁cnt,这是由于活性位点金属纳米颗粒的大微晶尺寸,这已经通过xrd技术被证实。此外,在ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂上形成的cnt属于基底生长模式。它用缠结的纤维挤满了密集的聚集。从sem图像中,我们可以观察到由于空间竞争,cnt在随机方向上生长。cnt的长度取决于工艺条件,尤其是停留时间。图44a-图44f示出大多数所形成的cnt的长度在50-200nm的范围内。观察到,所形成的cnt的壁是线性结构或弯曲结构,其中石墨烯层被倾斜地堆叠。在这些类型的cnt中,石墨层与cnt的轴线不一致。不希望受特定理论的束缚,当石墨烯层堆叠时,缺陷可能在边缘平面之间形成,这可以解释在拉曼光谱中观察到d带,但不能解释在tga分析中识别的非晶相。
对于60wt%的co/al2o3气凝胶催化剂和70wt%的co/al2o3气凝胶催化剂,如图45a-图45f中所示出的,具有50-150nm直径的多壁cnt与位于底部处的过渡金属纳米颗粒一起形成。可以观察到,这些cnt的壁具有平行的形态。与在ni/al2o3气凝胶催化剂上形成的cnt相比,它相对薄且直径较小。然而,对于那些通过常规初湿含浸技术合成的催化剂,所形成的cnt在尖端和基底处具有金属纳米颗粒。这些传统的催化剂提供了尖端生长的cnt和基底生长cnt的混合物(参见wangg等人,energy&fuels2013;27:4448-56)。通过不同方法制备的催化剂的生长模式是不同的。与常规催化剂相比,气凝胶催化剂提供了实现在常规催化剂中所缺乏的cnt的基底生长的选择性途径。
从本文讨论的tem图像可以观察到,在ni/al2o3气凝胶催化剂或co/al2o3气凝胶催化剂上形成的cnt基本上都是经由基底生长形成的。不希望受特定理论的束缚,这可能归因于金属纳米颗粒和气凝胶载体之间的强相互作用。此外,不希望受特定理论的束缚,在甲烷分解反应的初始阶段期间,碳扩散可以发生在牢固地锚定在气凝胶结构中的ni或co上,从而为碳渗透到载体和金属之间提供屏障。结果,在金属的外表面上的碳沉淀可以成为主导。cnt只能从被锚定在载体上的金属的上表面生长。不希望受特定理论的束缚,据信使用气凝胶催化剂形成的石墨烯平面强烈地化学吸附在金属表面上,降低金属纳米颗粒的表面能并且增强金属-载体的相互作用。此外,tem分析示出,石墨帽在金属纳米颗粒上形成,其在随后的碳碎片的沉积期间被向上提起。结果表明,cnt远离金属纳米颗粒生长,形成cnt的基底生长。
ni/al2o3气凝胶催化剂的再生试验。气凝胶催化剂再生试验使用60wt%的ni/al2o3气凝胶催化剂进行。基底生长的cnt的形成通过tem分析被证实。在t=650℃、tos=60min和ghsv=42000h-1的条件下进行ch4分解的第一循环之后,所使用的气凝胶催化剂在650℃使用10%的o2再生持续45分钟。等式1表示甲烷分解,而等式2表示氧化再生过程。整个过程重复五次,并且结果在图46a和图46b中示出。
ch4→ccnt+2h2(等式1)
选择60wt%的ni/al2o3气凝胶催化剂以进行再生试验。如图46a中所图示的,该气凝胶催化剂在最初的15min内示出67.2%的ch4转化率,并且在第二循环中逐渐失活至61.4%的ch4转化率。在第三循环中,该气凝胶催化剂在最初的15min内呈现出69.3%的相对较高的转化率,并且然后在最后30min内失活至44.7%的ch4转化率。在第四循环中,它开始于在最初的15min内的68.3%的ch4转化率,并且逐渐失活至39.7%的ch4转化率。在四个循环之后,60wt%的ni/al2o3气凝胶催化剂仍然可以恢复初始活性,在最初的15min内呈现出68.7%的ch4转化率,并且然后失活至34.8%的ch4转化率。基本上,在本研究中开发的再生方案能够恢复初始活性,并且需要进一步研究循环之间的ch4转化率的损失。如图46b中所示出的,发现在第二循环和第三循环中,对于该气凝胶催化剂,h2收率在44.3%-35.9%的范围内。对于第四循环和第五循环,h2收率下降到34.5%-26.7%的范围。
如图47a-图47d中所示出的,在第二循环中形成的cnt使用tem来表征,以证实cnt的基底生长。观察到,金属纳米颗粒保留在载体上,而cnt从基底生长。即使在第五循环之后,tem分析示出所形成的cnt仍然是基底生长的,如图48a-图48d中所示出的。本文的数据示出,所公开的气凝胶催化剂的改善的孔隙率和扭曲度具有重要意义,并且提供了增强的催化活性、稳定性和再现性。
总之,结果示出,与通过初湿含浸技术制备的单金属和双金属ni、fe和co常规催化剂相比,通过所公开的溶胶-凝胶方法合成的气凝胶催化剂呈现出增强的性能。不希望受特定理论的束缚,所公开的气凝胶催化剂的增强的性能可以归因于在气凝胶合成工艺期间在金属和载体的界面处发生的协同效应。此外,不希望受特定理论的束缚,可能的是,在再生后观察到的催化活性的微小变化可以归因于金属颗粒的烧结,或者需要进一步优化再生方案。例如,60wt%的ni/al2o3气凝胶催化剂示出79.2%的高ch4转化率。在co/al2o3气凝胶催化剂中,对甲烷转化的催化活性低于ni/al2o3气凝胶催化剂的催化活性。ni/al2o3气凝胶催化剂和co/al2o3气凝胶催化剂两者都产生基底生长的cnt,这被认为是在不牺牲催化剂位点的情况下易于收获的关键。在五个循环后,60wt%气凝胶催化剂仍呈现出68.7%的高ch4转化率,而同时继续提供基本上仅基底生长的cnt。据信,所公开的气凝胶催化剂包含增加数量的活性位点,同时呈现出相对强的金属-载体相互作用,从而导致基底生长的cnt的形成。所公开的气凝胶催化剂提供了一种基本上仅提供基底生长的cnt的新方法。
对于本领域技术人员来说将明显的是,可以在本公开内容中进行多种改变和变化而不偏离本公开内容的范围或精神。通过考虑本文公开的公开内容的说明书和实践,本公开内容的其他实施方案对本领域技术人员来说将是明显的。意图的是说明书和实施例仅被认为是示例性的,其中本公开内容的真实范围和精神由所附的权利要求指示。
- 还没有人留言评论。精彩留言会获得点赞!