分子筛催化剂及其制备方法和应用与流程
[0001]
本发明涉及硅分子筛领域,具体涉及一种分子筛催化剂及其制备方法和应用。
背景技术:
[0002]
silicalite-1分子筛又称全硅、纯硅分子筛,于1978年由美国联合碳化物公司的e.m.flanigen等首次被成功合成出来,属于“pentasil”家族的成员之一。全硅分子筛是一种具有mfi拓扑学结构的无铝分子筛,是zsm-5型结构分子筛家族中组成最简单的一种分子筛,其骨架仅含有硅原子和氧原子,基本结构单元为sio4四面体。mfi拓扑学结构的全硅分子筛拥有丰富的微孔结构和规整均匀的三维细孔道,具有确定的zsm-5型分子筛的晶体结构,较高的内比表面积,良好的热稳定性、吸附和脱附能力等性能。全硅分子筛在膜吸附分离、净化、催化材料等领域的开发应用正受到人们的日益重视。
[0003]
全硅分子筛的合成方法一般采用传统的有机原料水热法,硅源多选用固体氧化硅、硅溶胶、白炭黑或正硅酸乙酯(简称teos)等,模板剂多选用四丙基氢氧化铵(简称tpaoh)、低碳烃类季铵盐或胺类化合物等,在170℃的温度下晶化三天。美国联合碳化物公司(ucc)、瑞典stety和印度p.ratnasamy等研究小组曾在这方面开展过研究。他们将全硅分子筛主要应用于无机微孔材料研究领域。
[0004]
jp59164617a中公开的mfi结构的全硅分子筛,是以正硅酸乙酯(teos)为硅源,四丙基氢氧化铵为模板剂制备的。在catal.rev.-sci.eng.,39(4),395~424(1997)中的研究表明,以正硅酸乙酯为硅源合成的全硅分子筛具有较高的bet总比表面和外表面积,可分别达到400米2/克和15-30米2/克,且环己酮肟的转化率和己内酰胺的选择性与外表面积的增加成正比。
[0005]
cn102050464a中合成过程包括下列步骤:(1)将正硅酸乙酯与四丙基氢氧化铵在室温下混合、搅拌、充分水解3-5小时,补加水,形成摩尔浓度为tpaoh/sio2=0.2,h2o/sio2=20的混合物;(2)将上述混合物在密闭反应釜中,自生压力下100℃晶化3天,然后洗涤、过滤、干燥,将所得全硅分子筛原粉在550℃焙烧6小时,样品的bet比表面积为439米2/克、外比表面为60米2/克;(3)将一定目数的分子筛原料与碱性硅溶胶置于转盘式成型机中进行滚动成型,得到分子筛含量86%的球形全硅分子筛;(4)将球形全硅分子筛与碱性缓冲溶液在带压反应釜搅拌,然后洗涤、过滤、干燥,得到催化剂的压碎强度为2.2kg/颗粒。
[0006]
美国专利申请us4061724a披露了一种全硅分子筛的合成方法,以sio2含量为30重量%的水溶胶为硅源、naoh为碱源和四丙基溴化铵为模板剂,分子筛晶化前凝胶混合物摩尔组成为:4.1na2o:50sio2:691h2o:1tpabr,200℃晶化3天制得压碎强度为2.1kg/颗粒的催化剂。该方法所得到的分子筛用于环己酮肟气相贝克曼重排反应制备己内酰胺时,具有很高的环己酮肟转化率和己内酰胺选择性。
[0007]
由于mfi拓扑学结构全硅分子筛在挤条成型、压片成型,甚至滚动成型等方面存在很大困难,即使成型后,催化剂的压碎强度很不理想(<60n/cm或<1kg/颗粒),根本无法实现工业化应用。
[0008]
己内酰胺是生产锦纶、工业帘子线以及尼龙工程塑料三大系列产品的主要原料,其需求较高。所述己内酰胺一般通过环己酮肟的贝克曼重排反应来得到。目前,工业上通常采用以浓硫酸或发烟硫酸为催化剂的液相重排工艺。该工艺生产的己内酰胺占世界己内酰胺生产总量的90%左右,但是该工艺需要消耗大量的硫酸和氨水,一般每生产1吨己内酰胺将副产1.3-1.8吨硫酸铵,生产成本较高。另外硫酸的使用会造成设备腐蚀和环境污染等问题。
[0009]
固体酸催化剂上的环己酮肟气相贝克曼重排反应是实现己内酰胺无硫铵化的新工艺,具有无设备腐蚀、无环境污染等问题,产物的分离提纯也将大大简化,因此无硫铵化的气相贝克曼重排反应工艺受到业内人士的极大关注。
[0010]
为了研制适用于气相贝克曼重排反应的固体酸催化剂,国内外研究者已对氧化物(复合氧化物)、沸石分子筛等催化剂进行了大量的研究,结果表明大多数催化剂均具有一定的活性,但共同的缺点是催化剂容易失活,催化剂寿命短,不能达到工业化的要求。
[0011]
在气相法贝克曼重排反应中作为催化剂的固体酸有多种,如:二氧化硅-氧化铝催化剂、固体磷酸催化剂、含硼酸的催化剂、高硅/铝比mfi结构分子筛催化剂等。中国专利申请cn1256967a中披露了一种用于环己酮肟转化为己内酰胺反应的、含有mfi结构分子筛催化剂的制备方法。该方法的基本出发点是以酸性硅胶为粘结剂,其具体方法是:将烷氧基硅酸性水解制得的硅质低聚物与ph≤5的mfi结构分子筛的亚微颗粒的水或醇-水分散液混合,使混合物乳化、固化、洗涤、焙烧制得凝胶微球。
[0012]
目前,环己酮肟气相贝克曼重排反应的固定床或移动床工艺存在催化剂寿命短,难以长周期连续运行,氮肟摩尔比高,移热困难,技术经济性差等缺点。
技术实现要素:
[0013]
本发明的目的是为了克服现有技术存在的上述缺陷,提供一种分子筛催化剂的制备方法和由该制备方法制得的分子筛催化剂以及该催化剂在环己酮肟气相贝克曼重排反应中的应用,本发明提供的分子筛催化剂制备方法制得的催化剂用于环己酮肟气相贝克曼重排反应过程中,可以提高环己酮肟的转化率和己内酰胺的选择性,具有较长使用寿命,提升环己酮肟气相贝克曼重排新工艺技术的经济性。
[0014]
本发明第一方面提供一种分子筛催化剂的制备方法,该方法包括:
[0015]
(1)将硅源、有机胺、有机模板剂、金属源、有机醇和水混合,得到胶体混合物,其中,硅源、有机胺、有机模板剂、有机醇和水的用量摩尔比为1:(0.05-0.5):(0.05-0.5):(4-20):(5-100),硅源与金属源的用量质量比为(10000-200000):1,硅源以sio2计,金属源以金属元素计;
[0016]
(2)将所述胶体混合物进行两段变温醇-水热体系晶化,所述两段变温醇-水热体系晶化的条件包括:在40-70℃下晶化0.5-5天,然后在80-130℃下晶化0.5-5天;
[0017]
(3)将步骤(2)得到的晶化母液进行过滤、洗涤、干燥,得到分子筛原粉;
[0018]
(4)将步骤(3)得到的分子筛原粉进行粉碎、滚动成型、焙烧和含氮化合物后处理;
[0019]
所述金属选自过渡金属和iiia族金属中的至少一种。
[0020]
优选地,所述有机胺为三正丙胺。
[0021]
优选地,所述两段变温醇-水热体系晶化的条件包括:在50-65℃下晶化1-1.5天,
然后在100-120℃下晶化1.5-2天。
[0022]
本发明第二方面提供上述制备方法制得的分子筛催化剂。
[0023]
本发明第三方面提供上述分子筛催化剂在环己酮肟气相贝克曼重排反应中的应用。
[0024]
本发明提供的催化剂的制备方法中,在分子筛合成过程中额外加入醇和金属源,并采用有机胺和有机模板剂配合使用,且采用两段变温醇-水热体系晶化,非常有利于微量的金属元素进入到分子筛骨架,将分子筛原粉经粉碎、滚动成型处理、焙烧和含氮化合物后处理得到的催化剂催化性能好。本发明制备的分子筛催化剂中含有微量金属元素,所述金属优选选自al、ag、co、ni、cu、zn、mn、pd、pt、cr、fe、la、au、ru、rh、y、ce、pt、rh、ti、zr、v、mo和w中的至少一种。采用上述优选的金属元素更有利于提高催化剂的转化率和选择性。
[0025]
与现有技术对比,本发明的有益效果包括:在环己酮肟气相贝克曼重排反应上,采用现有全硅分子筛作催化剂,环己酮肟转化率和己内酰胺选择性均比较高,快速评价第6小时分别达到96%和94.5%,基本上达到极限,但随着反应时间的延长,很难保证催化剂稳定性和寿命。采用本发明的制备方法制得了高结晶度、细颗粒细的接近中性的全硅分子筛,将其进行滚动成型后所得的球形催化剂压碎强度好,在移动床或固定床反应体系中,以该球形全硅分子筛为催化剂进行环己酮肟气相贝克曼重排反应制备己内酰胺的方法,能实现己内酰胺长周期、连续生产,在保持己内酰胺选择性基本不变的情况下,可以提高环己酮肟的转化率,延长催化剂寿命,例如,本发明提供催化剂压碎强度可达2.5kg/颗粒以上,将该催化剂用于环己酮肟气相贝克曼重排反应过程中,催化剂在运转6h时,环己酮肟转化率可以达到97%以上,运转600h时,环己酮肟转化率可以达到99%以上。
附图说明
[0026]
图1是本发明实施例1所制得的催化剂的形貌照片。
具体实施方式
[0027]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0028]
本发明所述“第一”和“第二”不起到限定作用,只是为了区分不同阶段进行的操作或者不同阶段加入的物料。
[0029]
本发明第一方面提供一种分子筛催化剂的制备方法,该方法包括:
[0030]
(1)将硅源、有机胺、有机模板剂、金属源、有机醇和水混合,得到胶体混合物,其中,硅源、有机胺、有机模板剂、有机醇和水的用量摩尔比为1:(0.05-0.5):(0.05-0.5):(4-20):(5-100),硅源与金属源的用量质量比为(10000-200000):1,硅源以sio2计,金属源以金属元素计;
[0031]
(2)将所述胶体混合物进行两段变温醇-水热体系晶化,所述两段变温醇-水热体系晶化的条件包括:在40-70℃下晶化0.5-5天,然后在80-130℃下晶化0.5-5天;
[0032]
(3)将步骤(2)得到的晶化母液进行过滤、洗涤、干燥,得到分子筛原粉;
[0033]
(4)将步骤(3)得到的所述分子筛原粉进行粉碎、滚动成型、焙烧和含氮化合物后处理;
[0034]
所述金属选自过渡金属和iiia族金属中的至少一种。
[0035]
在本发明中,在无特殊说明情况下,所述用量摩尔比和用量质量比指的是物料进料(投料)时的用量摩尔比和用量质量比。
[0036]
根据本发明提供的方法,优选地,所述硅源为有机硅源,进一步优选为有机硅酸酯,例如可以为通式是(or1)4si的有机硅酸酯,其中,r1为c1-c4的烷基。
[0037]
根据本发明提供的方法,最优选地,所述硅源为正硅酸乙酯和/或正硅酸甲酯。
[0038]
根据本发明提供的方法,优选地,所述有机胺选自脂肪胺类化合物中的至少一种。具体地,所述的脂肪胺类化合物的通式可以为(r2)
k
(nh
3-k
)
n
,r2为具有1-6个碳原子的烷基,n=1或2,k=1、2或,3;进一步优选地,所述脂肪胺类化合物可以选自乙胺、一正丙胺、二正丙胺、三正丙胺、正丁胺、乙二胺和己二胺中的至少一种,最优选为三正丙胺。
[0039]
根据本发明提供的方法,优选地,所述有机模板剂选自季铵碱类化合物,进一步优选为四丙基氢氧化铵和/或四乙基氢氧化铵。所述季铵碱类化合物可以为含有1-4个碳原子的烷基季铵碱类化合物,进一步优选为四丙基氢氧化铵和/或四乙基氢氧化铵。
[0040]
根据本发明提供的方法,所述金属可以选自过渡金属和iiia族金属中的至少一种。进一步地,所述过渡金属选自第ib族、第iib族、第ivb族、第vb族、第vib族、第viib族和第viii族金属中的至少一种。
[0041]
根据本发明提供的方法,优选地,所述金属选自al、ag、co、ni、cu、zn、mn、pd、pt、cr、fe、la、au、ru、rh、y、ce、pt、rh、ti、zr、v、mo和w中的至少一种,进一步优选地,所述金属选自fe、ni、ti、pd、ce、al、cu、zr、pt和la中的至少一种,最优选为fe、ti、ce、al、zr和pt中的至少一种。采用该种优选实施方式,更有利于提高催化剂的催化性能。
[0042]
根据本发明提供的方法,所述金属源为能够提供上述金属的各种含金属元素的化合物,优选地,所述金属源选自金属的氧化物、硝酸盐、氯化物、硫酸盐、醋酸盐和酯类金属化合物中的至少一种。所述酯类金属化合物可以为钛酸乙酯和/或钛酸丁酯。
[0043]
根据本发明提供的方法,所述金属源优选为fe(no3)3、钛酸四丁酯、ce(no3)4、al(no3)3、zrocl2和h2ptcl6中的至少一种。上述金属源可以含有结晶水,也可以没有,本发明对此没有特别的限定。
[0044]
根据本发明提供的方法,所述有机醇可以为c1-c4的一元醇、二元醇中的至少一种,优选地,所述有机醇为甲醇和/或乙醇。
[0045]
本发明对步骤(1)所述混合的具体实施方式没有特别的限定,只要能够得到所述胶体混合物即可。
[0046]
根据本发明的一种优选方式,所述硅源为正硅酸乙酯,所述有机醇为乙醇。本发明的发明人在研究过程中发现,正硅酸乙酯作为硅源和乙醇作为有机醇配合使用更有利于进一步提高制得的催化剂的催化性能。进一步优选地,步骤(1)所述混合包括:将乙醇、有机胺和有机模板剂进行第一混合,然后加入金属源和水,再加入正硅酸乙酯;或者,步骤(1)所述混合包括:将乙醇、有机胺和有机模板剂进行第一混合,然后依次加入水和正硅酸乙酯,再加入金属源。该种优选实施方式更有利于各物料的混合,同时更有利于发挥各物料的配合作用。
[0047]
根据本发明的一种具体实施方式,所述第一混合在搅拌条件下进行,对于搅拌的时间没有特别的限定,只要使得乙醇、有机胺和有机模板剂混合均匀即可。具体地,该方法还可以包括,在加入金属源和水之后,进行搅拌,然后加入所述正硅酸乙酯。
[0048]
根据本发明的一种具体实施方式,该方法还可以包括:在加入正硅酸乙酯之后,进行搅拌以得到所述胶体混合物。本发明对所述搅拌的条件没有特别的限定,以能够得到所述胶体混合物为准,例如,可以在10-50℃下搅拌0.5-10小时。
[0049]
根据本发明的另一种优选实施方式,所述硅源为正硅酸甲酯,所述有机醇为甲醇。本发明的发明人在研究过程中发现,正硅酸甲酯作为硅源和甲醇作为有机醇配合使用更有利于进一步提高制得的催化剂的催化性能。优选地,步骤(1)所述混合包括:将甲醇、有机胺和有机模板剂进行第二混合,然后加入金属源和水,再加入正硅酸甲酯;进一步优选地,所述正硅酸甲酯通过多次加入,更优选在10-30℃恒温下进行。采用该种优选实施方式,更有利于控制正硅酸甲酯的水解速度,进而使得制得的催化剂的催化性能能够得到进一步的提高。
[0050]
本发明对所述正硅酸甲酯通过多次加入的具体操作没有特别的限定,具体地,可以将所需量的正硅酸甲酯分为相等或不等的多份(优选为3-10份),然后将各份正硅酸甲酯间隔加入。本发明对所述间隔的时间没有特别的限定,所述间隔时间可以根据每次加入的正硅酸甲酯的量考虑,加入的正硅酸甲酯的量多,间隔时间可以适当延长,加入的正硅酸甲酯的量少,间隔时间可以适当地缩短。优选地,间隔时间可以为5-30min,各间隔时间可以相等也可以不等。本发明实施例中以正硅酸甲酯分为4批次,间隔时间为10min为例进行示例性说明,本发明并不限于此。
[0051]
根据本发明的一种优选实施方式,硅源、有机胺、有机模板剂、有机醇和水的用量摩尔比为1:(0.05-0.3):(0.05-0.3):(4-15):(15-50),进一步优选为1:(0.1-0.3):(0.1-0.2):(6-13):(20-40)。
[0052]
根据本发明,优选地,硅源与金属源的用量质量比为(12000-140000):1,更优选为(14000-50000):1。采用该种优选实施方式,进入分子筛骨架的适宜量的金属,更有利于提高催化剂的催化性能。
[0053]
根据本发明提供的方法,优选地,所述两段变温醇-水热体系晶化的条件包括:在50-65℃下晶化1-1.5天,然后在100-120℃下晶化1.5-2天。采用该种优选的水热体系晶化条件制得的催化剂具有更好的催化性能。
[0054]
具体地,所述两段变温醇-水热体系晶化可以在密闭体系下,在自生压力下进行,例如在密闭反应釜中进行。
[0055]
根据本发明的一种优选实施方式,该方法还包括:在步骤(3)所述过滤之前,将所述晶化母液进行赶醇。优选地,所述赶醇的条件包括:温度为50-85℃,时间为1-12h。
[0056]
根据本发明提供的方法,所述过滤和洗涤可以为本领域常规使用的各种过滤和洗涤方法,对二者的顺序也没有特别的限定。本发明对所述洗涤过程使用的洗涤用剂没有特别的限定,例如可以为水。优选地,所述洗涤洗至过滤出来的洗涤水ph为7.5-10,优选为8-9。
[0057]
根据本发明的提供的方法,对干燥的条件选择范围较宽,例如所述干燥的条件可以包括:干燥温度为80-150℃,干燥时间为2-36h;优选地,干燥温度为100-120℃,干燥时间
10-30小时。
[0058]
根据本发明提供的方法,具体地,步骤(4)中,将所述分子筛原粉粉碎后与粘结剂混合,然后进行所述滚动成型。优选地,所述分子筛原粉粉碎后得到的固体物质与粘结剂的质量用量比为1:(0.002-0.25),更优选为1:(0.02-0.15)。
[0059]
根据本发明提供的方法,加入粘结剂的目的是为了使粉体粒子在转动时互相粘结在一起,可以进一步提高成型产品的强度。
[0060]
所述滚动成型可以是一步成型,也可以是分步骤成型,优选为分步骤成型。
[0061]
根据本发明的一种优选实施方式,步骤(4)所述滚动成型包括:
[0062]
a.从粉碎得到的固体物质中选取颗粒大小为100-1000目的第一部分分子筛,将第一部分分子筛与第一粘结剂混合,进行第一滚动成型,得到颗粒大小为0.1-0.8mm的第一颗粒,其中,第一部分分子筛与第一粘结剂的质量比为1:(0.2-1);
[0063]
b.从粉碎得到的固体物质中选取颗粒大小为100-1000目的第二部分分子筛,将第二部分分子筛、第二粘结剂和第一颗粒混合,进行第二滚动成型,得到颗粒大小为1.3-2.5mm的第二颗粒,其中,第二部分分子筛与第二粘结剂的质量比为1:(0.001-0.5)。
[0064]
在上述优选实施方式中,选取颗粒大小为100-1000目的部分分子筛可以通过筛分的方式进行。所述颗粒大小指的是颗粒上的任意两个不同点之间的最大直线距离,当所述颗粒为球形时,所述颗粒大小指其直径。
[0065]
优选地,所述第一颗粒和第二颗粒各自独立地为球形。
[0066]
根据本发明,优选地,步骤b中的所述第二部分分子筛与第二粘结剂可以分别加入以与第一颗粒混合,还可以经预先混合均匀后与第一颗粒混合,优选将第二部分分子筛与第二粘结剂预先混合均匀后与第一颗粒混合。
[0067]
进一步优选地,步骤b中所述将第二部分分子筛、第二粘结剂和第一颗粒混合包括:将第二部分分子筛与第二粘结剂混合,粉碎,取30目以下的颗粒与第一颗粒混合。采用该种优选实施方式更有利于进一步提高催化剂的压碎强度。
[0068]
根据本发明,第一部分分子筛与第二部分分子筛的重量比可以为根据实际需要的任意比例,也可以根据分子筛成球的情况随时进行调整,本发明不做特别的限制。
[0069]
根据本发明,所述滚动成型优选在转盘成型机中进行。上述第一滚动成型和第二滚动成型可以是在同一个转盘成型机中进行,也可以在两个转盘成型机中进行,优选在同一个转盘成型机中进行。
[0070]
根据本发明提供的方法,本发明的发明人在研究过程中发现,转盘滚球成型的停留时间、转盘倾角、转盘直径d、转盘深度h、转盘转速n均可能会对滚动成型产生影响。其中,所述停留时间是指一定目数的分子筛原料从加入转盘成型机到形成目标颗粒、脱离转盘成型机的平均时间,通常可以为10-600分钟、优选为30-180分钟;所述转盘倾角是指转盘与水平线的夹角,可以为40-55
°
,优选为45-50
°
,在小于40
°
时,成型状态不好,倾角越大,球的尺寸越小;所述转盘直径d与转盘深度h之间的关系优选为h=0.1-0.25d;转盘转速优选为10-50rpm,进一步优选为20-40rpm。
[0071]
根据本发明提供的方法,转盘成型机的处理量以每小时生产催化剂的量计可以为20-100kg/h,例如为60kg/h;转盘中的存料量优选为处理量的1/10-1/4。所述存料量是指转盘中未达到合格直径的微、小球催化剂的量。采用该种方法更有利于使得成型产品获得更
好的机械强度及形态保存性,避免产品颗粒分层脱皮。
[0072]
根据本发明,优选地,所述粘结剂为水和/或硅溶胶。所述硅溶胶可以是酸性硅溶胶,也可以是碱性硅溶胶,可以通过商购得到,也可以按照任意一种现有技术制备得到,进一步优选地,所述硅溶胶为碱性硅溶胶。
[0073]
优选地,所述硅溶胶中,sio2含量为20-45重量%。
[0074]
优选地,所述硅溶胶的钠离子含量为10-300ppm。需要说明的是,钠离子在催化剂制备过程中的水洗步骤基本上会被洗掉,催化剂上大概会残存20-30ppm。
[0075]
根据本发明的一种优选实施方式,该方法还包括:在所述滚动成型之后,在焙烧之前,将滚动成型得到的产物进行抛光处理。采用该种优选实施方式一方面可以增加球形催化剂外表面的圆整度,另一方面可以进一步增加催化剂的压缩强度。所述抛光处理可以按照本领域常规技术手段进行。例如,将滚动成型得到的产物在20-50℃下吹风(可以赶走水分),吹风过程中多次(例如3-10次)补极其微量水(可以使得催化剂表面润湿,容易轻微小范围的变形,提高球的圆整度),然后进行收紧(不补水仅吹风,一般可以进行1-4小时)。
[0076]
根据本发明的一种具体实施方式,该方法还包括在所述焙烧之前,对滚动成型产物进行干燥。所述干燥的条件可以包括:干燥温度为80-150℃,干燥时间为2-36h;优选地,干燥温度为100-120℃,干燥时间10-30小时。
[0077]
本发明步骤(4)所述焙烧可以在常规加热炉中进行,例如可以在加热梭式炉中进行,优选地,步骤(4)所述焙烧的条件包括:温度为400-600℃,时间为1-24h,进一步优选地,温度为400-550℃,时间为1-10h。
[0078]
根据本发明的一种优选实施方式,所述含氮化合物后处理包括:将焙烧得到的焙烧产物与含氮化合物的碱性缓冲溶液接触,然后进行干燥。采用该种优选实施方式更有利于提供制得的催化剂的催化性能。
[0079]
根据本发明,优选地,所述含氮化合物的碱性缓冲溶液含有铵盐和碱,其溶剂可以为水。所述含氮化合物可以为铵盐,例如可以为硝酸铵和/或醋酸铵。所述碱可以选自氨水、四甲基氢氧化铵、四乙基氢氧化铵和四丙基氢氧化铵中的至少一种,优选为氨水。
[0080]
根据本发明的一种优选实施方式,所述铵盐的含量为0.5-20重量%;所述碱的含量为5-30重量%。本发明实施例中铵盐含量以7.5重量%、碱的含量以26重量%为例,进行示例性说明,本发明并不限于此。
[0081]
优选地,所述含氮化合物的碱性缓冲溶液的ph值为8.5-13.5,进一步优选为9-12,更优选为11-11.5。
[0082]
优选地,所述焙烧产物与含氮化合物的碱性缓冲溶液的重量比为1:(5-15)。
[0083]
优选地,所述接触的条件包括:温度为50-120℃,压力为0.5-5kg/cm2,时间为10-300min。进一步优选地,所述接触在搅拌条件下进行。本发明对所述搅拌的速度没有特别的限定,本领域技术人员可以根据实际情况进行适当的选择。
[0084]
根据本发明提供的方法,与含氮化合物的碱性缓冲溶液接触的过程可以进行重复操作。对于重复的次数本发明不做特别的限定,可以根据催化剂的性能确定,例如可以重复1-3次。
[0085]
本发明对所述含氮化合物后处理中干燥的条件没有特别的限定,可以按照本领域常规技术手段进行,所述干燥只要将水分除去即可,所述干燥的方法包括但不限于自然干
燥、加热干燥、鼓风干燥,所述干燥的温度可以为100-120℃,干燥的时间可以为10-24小时。
[0086]
根据本发明,具体地,该方法还可以包括:在所述干燥之前,将所述焙烧产物与含氮化合物的碱性缓冲溶液接触之后得到的物质依次进行过滤、洗涤。本发明对所述洗涤过程使用的洗涤用剂没有特别的限定,例如可以为水。
[0087]
本发明第二方面提供上述的制备方法制得的分子筛催化剂,该催化剂的压碎强度σ>2.0kg/颗粒,优选为>2.5kg/颗粒,进一步优选为2.5-2.8kg/颗粒。
[0088]
本发明第三方面提供上述分子筛催化剂在环己酮肟气相贝克曼重排反应中的应用。将本发明提供的分子筛催化剂用于环己酮肟气相贝克曼重排反应,可以提高环己酮肟的转化率和已内酰胺的选择性,且可以延长催化剂寿命,提升气相重排新工艺技术的经济性。
[0089]
根据本发明提供的应用,可以是将环己酮肟在溶剂存在下与上述催化剂接触以进行气相贝克曼重排反应。将本发明的分子筛催化剂应用于环己酮肟气相贝克曼重排反应时,环己酮肟转化率和己内酰胺的选择性较高,能实现己内酰胺长周期、连续生产,比现有全硅分子筛催化剂具有更高的己内酰胺选择性和收率。并且由于副产物总量下降,使得产物分离能耗随之下降,技术经济性得到有效提高。
[0090]
优选地,所述溶剂与环己酮肟的摩尔比为(2-10):1。所述溶剂可以选自c1-c6的脂肪醇,优选为甲醇、乙醇和正丙醇中的至少一种。
[0091]
优选地,所述气相贝克曼重排反应在氮气的存在下进行,所述氮气与环己酮肟的摩尔比为(10-80):1,进一步优选为(40-60):1。
[0092]
优选地,所述气相贝克曼重排反应的条件包括:环己酮肟的重量空速(whsv)为0.1-20小时-1
,优选为0.5-16小时-1
;反应温度为300-500℃,优选为350-400℃,更优选为360-390℃;以表压计,反应压力为0.1-0.5mpa。
[0093]
以下通过实施例对本发明进行详细描述。
[0094]
金属元素含量使用美国pe(珀金埃尔默)公司7000dv型icp电感耦合等离子体原子发射光谱仪进行测定,测试条件为:用hf酸或王水溶解分子筛,使样品中的氧化硅和金属氧化物彻底溶解,在水溶液中测定金属离子含量。
[0095]
压碎强度σ按照《石油化工分析方法》(杨翠定等人,科学出版社,1990年)中的ripp25-90方法在颗粒强度测定仪qcy-602型(原化工部制碱工业研究所生产)上测得。
[0096]
以下实施例中滚动成型在转盘成型机中进行,所述转盘式成型机为泰州市天泰制药机械厂生产的包衣机(糖衣机),型号为by-1200型。
[0097]
实施例1
[0098]
(1)向3m3不锈钢反应釜中分别加入870kg的95重量%乙醇(etoh)、58.5kg三正丙胺和180kg的22.5wt%的四丙基氢氧化铵(tpaoh)水溶液,搅拌,继续向反应釜中加入540kg水和37.2g的fe(no3)3·
9h2o,继续搅拌,最后再加入416kg正硅酸乙酯,常温(25℃)下充分搅拌6小时,形成胶体混合物,其中,硅源:(c3h8)3n:tpaoh:etoh:h2o的用量摩尔比=1:0.2:0.1:9:20,硅源与fe
3+
的用量质量比为23600:1,其中,硅源以sio2计。
[0099]
(2)将上述胶体混合物先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天。
[0100]
(3)将反应釜降温,打开釜盖,在75℃温度下进行6小时的赶乙醇处理,然后采用
50nm六管膜进行膜过滤并采用50℃的水进行洗涤,至洗涤水ph值达到或接近9.0,洗涤后得到的分子筛浆液在120℃干燥20小时,得到本实施例滚动成型所需的分子筛原粉。
[0101]
取少量上述得到的分子筛原粉在550℃焙烧6小时,得到的分子筛中,铁离子含量为42μg/g。该分子筛晶粒大小均匀,粒径为0.15-0.25μm。
[0102]
(4)将步骤(3)得到的分子筛原粉在粉碎机中粉碎,取2kg100-1000目的粉碎后分子筛原料置于转盘成型机中,所用转盘成型机的转盘直径1.2m,转盘深度为450mm,转盘倾角确定为50
°
,转盘转速设定30rpm。向其中喷洒去离子水1.5kg,得到直径大约0.2-0.8mm的第一球形颗粒。
[0103]
另取220kg 200-800目的粉碎后分子筛原料与100kg的sio2含量为30重量%的碱性硅溶胶混合均匀并重新粉碎,取用小于30目的颗粒向上述具有第一球形颗粒的转盘成型机中匀速加入300kg,240min内加完。其中,中途多次用12目和9目的筛子筛分,得到直径1.7-2.2mm第二球形颗粒,约160kg,小于1.7mm的球形颗粒倒回转盘成型机中继续长大。
[0104]
将上述得到的90kg第二球形颗粒在45℃下吹风,中途多次补极其微量水,收紧2小时,在120℃干燥24小时,之后在550℃下焙烧10小时。最终得到分子筛含量86%的焙烧产物。
[0105]
(5)将90kg上述焙烧产物与900kg碱性缓冲溶液(该碱性缓冲溶液为氨水和硝酸铵水溶液的混合液,其中,氨水的含量为26重量%,硝酸铵水溶液中硝酸铵的含量为7.5重量%,氨水与硝酸铵水溶液的重量比为3:2,碱性缓冲溶液的ph值为11.35)加入到带压反应釜中,在80℃、2.3kg/cm2压力下搅拌1小时,然后洗涤、过滤、干燥,得到催化剂编号为a1。该催化剂的形貌照片如图1所示。
[0106]
测得催化剂a1的压碎强度σ列于表1。
[0107]
对比例1
[0108]
按照实施例1的方法,不同的是,步骤(2)中,醇-水热体系晶化的条件为:100℃下醇-水热体系晶化3天。得到催化剂d1。
[0109]
对比例2
[0110]
按照实施例1的方法,不同的是,催化剂制备过程中不添加乙醇,即步骤(1)中不添加乙醇。得到催化剂d2。
[0111]
对比例3
[0112]
按照实施例1的方法,不同的是,催化剂制备过程中不添加fe(no3)3·
9h2o。得到催化剂d3。
[0113]
对比例4
[0114]
按照实施例1的方法,不同的是,催化剂制备过程中不添加三正丙胺。得到催化剂d4。
[0115]
对比例5
[0116]
本对比例说明按照中国专利申请cn1338427a的方法二合成全硅分子筛的过程。
[0117]
在室温25℃下将278kg正硅酸乙酯倒入2m3不锈钢反应釜中,搅拌30分钟,240kg的22.5wt%四丙基氢氧化铵水溶液加入正硅酸乙酯中,室温25℃下搅拌水解5小时,加水296kg,加乙醇534kg,搅拌均匀为溶胶,此时混合溶胶的化学组成为h2o/sio2=20,etoh/sio2=12.7,tpaoh/sio2=0.20,在110℃晶化2天,过滤,洗涤,至洗涤水ph值达到或接近
9.0,洗涤后得到的分子筛浓缩浆液在120℃干燥20小时,得到本对比例滚动成型所需的分子筛原粉,分子筛原粉在粉碎机中粉碎。
[0118]
取220kg 200-800目的粉碎后分子筛原料与100kg的sio2含量为30重量%的碱性硅溶胶置于转盘成型机中进行滚动成型,所用转盘转动成型机的转盘直径1.2m,转盘深度为450mm,转盘倾角确定为50
°
,转盘转速设定30rpm,得到直径大约1.7-2.2mm的球形颗粒后在120℃干燥24小时,在550℃下焙烧10小时得到焙烧产物。
[0119]
将90kg上述焙烧产物与900kg碱性缓冲溶液(该碱性缓冲溶液为氨水和硝酸铵水溶液的混合液,其中,氨水的含量为26重量%,硝酸铵水溶液中硝酸铵的含量为7.5重量%,氨水与硝酸铵水溶液的重量比为3:2,碱性缓冲溶液的ph值为11.35)加入到带压反应釜(kcf-2型磁力搅拌高压釜,烟台高新区科立自控设备研究所)中,在80℃、2.3kg/cm2压力下搅拌1小时,然后洗涤、过滤、干燥,得到催化剂编号为d5。催化剂压碎强度σ列于表1。
[0120]
实施例2
[0121]
(1)向3m3不锈钢反应釜中分别加入580kg的95重量%乙醇、87.6kg三正丙胺和180kg的22.5wt%的四丙基氢氧化铵水溶液,搅拌,继续向反应釜中加入900kg水,搅拌,最后再加入416kg正硅酸乙酯,充分混合,最后加入35.6g钛酸四丁酯,常温(25℃)下充分搅拌6小时,形成ph值为13.39胶体混合物,其中,硅源:(c3h8)3n:tpaoh:etoh:h2o的用量摩尔比=1:0.3:0.1:6:30,硅源与ti
4+
的用量质量比为23600:1,其中,硅源以sio2计。
[0122]
(2)将上述胶体混合物先在50℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天。
[0123]
(3)将反应釜降温,打开釜盖,在75℃温度下进行6小时的赶乙醇处理,然后采用50nm六管膜进行膜过滤并采用50℃的水进行洗涤,至洗涤水ph值达到或接近9.0,洗涤后得到的分子筛浆液在120℃干燥20小时,得到本实施例滚动成型所需的分子筛原粉。
[0124]
取少量上述得到的分子筛原粉在550℃焙烧6小时,得到的分子筛中,测得钛离子含量为41.8μg/g。该分子筛晶粒大小均匀,粒径为0.15-0.25μm。
[0125]
(4)将步骤(3)得到的分子筛原粉在粉碎机中粉碎,取2kg100-1000目的粉碎后分子筛原料置于转盘成型机中,所用转盘成型机的转盘直径1.2m,转盘深度为450mm,转盘倾角确定为50
°
,转盘转速设定30rpm。向其中喷洒去离子水1.4kg,得到直径大约0.2-0.8mm的第一球形颗粒。
[0126]
另取200kg200-800目的粉碎后分子筛原料与40kg的sio2含量为30重量%的碱性硅溶胶混合,再向其中补加55kg水,均匀混合,并重新粉碎,取用小于30目的颗粒向上述具有第一球形颗粒的转盘成型机中匀速加入280kg,300min内加完。其中,中途多次用12目和9目的筛子筛分,得到直径1.7-2.2mm第二球形颗粒,约150kg,小于1.7mm的球形颗粒倒回转盘成型机中继续长大。
[0127]
将上述得到的100kg第二球形颗粒在45℃下吹风,中途多次补极其微量水,收紧2小时,在120℃干燥24小时,之后在550℃下焙烧10小时。最终得到分子筛含量93%的焙烧产物。
[0128]
(5)将90kg上述焙烧产物与900kg碱性缓冲溶液(该碱性缓冲溶液为氨水和硝酸铵水溶液的混合液,其中,氨水的含量为26重量%,硝酸铵水溶液中硝酸铵的含量为7.5重量%,氨水与硝酸铵水溶液的重量比为3:2,碱性缓冲溶液的ph值为11.35)加入到带压反应
釜中,在100℃、2.8kg/cm2压力下搅拌1小时,然后洗涤、过滤、干燥,得到催化剂编号为a2。
[0129]
测得催化剂a2的压碎强度σ列于表1。
[0130]
实施例3
[0131]
(1)向3m3不锈钢反应釜中分别加入870kg的95重量%乙醇、58.4kg三正丙胺和180kg的22.5wt%的四丙基氢氧化铵水溶液,搅拌,继续向反应釜中加入540kg水和19.2g ce(no3)4·
7h2o,继续搅拌,最后再加入416kg正硅酸乙酯,充分混合,常温下(25℃)充分搅拌6小时,形成ph值为12.49的胶体混合物,其中,硅源:(c3h8)3n:tpaoh:etoh:h2o的用量摩尔比=1:0.20:0.10:9:20,硅源与ce
4+
的用量质量比为23500:1,其中,硅源以sio2计。
[0132]
(2)将上述胶体混合物先在65℃下醇-水热体系晶化1天,再在120℃下醇-水热体系晶化2天。
[0133]
(3)将反应釜降温,打开釜盖,在75℃温度下进行6小时的赶乙醇处理,然后采用50nm六管膜进行膜过滤并采用50℃的水进行洗涤,至洗涤水ph值达到或接近9.0,洗涤后得到的分子筛浆液在120℃干燥20小时,得到本实施例滚动成型所需的分子筛原粉。
[0134]
取少量上述得到的分子筛原粉在550℃焙烧6小时,,得到的分子筛中,铈离子含量为42μg/g,该分子筛晶粒大小均匀,粒径为0.15-0.25μm。
[0135]
(4)将步骤(3)得到的分子筛原粉在粉碎机中粉碎,取2kg100-1000目的粉碎后分子筛原料置于转盘成型机中,所用转盘转动成型机的转盘直径1.2m,转盘深度为450mm,转盘倾角确定为50
°
,转盘转速设定30rpm。向其中加入sio2含量为30重量%的碱性硅溶胶1.5kg,得到直径大约0.2-0.8mm的第一球形颗粒。
[0136]
另取200kg 200-800目的粉碎后分子筛原料与50kg的sio2含量为40重量%的碱性硅溶胶混合均匀,再向其中补加45kg水,均匀混合,并重新粉碎,取用小于30目的颗粒向上述具有第一球形颗粒的转盘成型机中匀速加入280kg,300min内加完。其中,中途多次用12目和9目的筛子筛分,得到直径1.7-2.2mm第二球形颗粒,约150kg,小于1.7mm的球形颗粒倒回转盘成型机中继续长大。
[0137]
将上述得到的100kg第二球形颗粒在45℃下吹风,中途多次补极其微量水,收紧2小时,在120℃干燥24小时,之后在550℃下焙烧10小时。最终得到分子筛含量89.5%的焙烧产物。
[0138]
(5)将90kg上述焙烧产物与900kg碱性缓冲溶液(该碱性缓冲溶液为氨水和硝酸铵水溶液的混合液,其中,氨水的含量为26重量%,硝酸铵水溶液中硝酸铵的含量为7.5重量%,氨水与硝酸铵水溶液的重量比为3:2,碱性缓冲溶液的ph值为11.35)加入到带压反应釜中,在80℃、2.3kg/cm2压力下搅拌3小时,然后洗涤、过滤、干燥,得到催化剂编号为a3。
[0139]
测得催化剂a3的压碎强度σ列于表1。
[0140]
实施例4
[0141]
(1)在25℃恒温下分别将575kg甲醇(meoh)、58.5kg三正丙胺和180kg22.5%的四丙基氢氧化铵水溶液加入到3m3不锈钢反应釜中,搅拌,继续向反应釜中加入580kg水和69.6g al(no3)3·
9h2o,搅拌30分钟,分四批次、每批次间隔15分钟,将304kg正硅酸甲酯加入反应釜中,搅拌60分钟,之后在25℃恒温下继续搅拌水解2小时,搅拌均匀,形成ph值为12.58的胶体混合物,其中,硅源:(c3h8)3n:tpaoh:meoh:h2o的用量摩尔比=1:0.20:0.10:9:20,硅源与al
3+
的用量质量比为23600:1,其中,硅源以sio2计。
[0142]
(2)将上述胶体混合物先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天。
[0143]
(3)将反应釜降温,打开釜盖,在75℃温度下进行6小时的赶甲醇处理,然后采用50nm六管膜进行膜过滤并采用50℃的水进行洗涤,至洗涤水ph值达到或接近9.0,洗涤后得到的分子筛浆液在120℃干燥20小时,得到本实施例滚动成型所需的分子筛原粉。
[0144]
取少量上述得到的分子筛原粉在550℃焙烧6小时,得到的分子筛中,铝离子含量为41μg/g。该分子筛晶粒大小均匀,粒径为0.15-0.25μm。
[0145]
(4)将步骤(3)得到的分子筛原粉在粉碎机中粉碎,取10kg200-500目的粉碎后分子筛原料置于转盘成型机中,所用转盘滚动成型机的转盘直径1.2m,转盘深度为450mm,转盘倾角确定为50
°
,转盘转速设定30rpm。向其中喷洒去离子水5.8kg左右,得到直径大约0.2-0.8mm的第一球形颗粒。
[0146]
另取200kg 200-800目的粉碎后分子筛原料与95kg去离子水混合均匀后向上述具有第一球形颗粒的转盘成型机中匀速加入,300min内加完。其中,中途多次用12目和9目的筛子筛分,得到直径1.7-2.2mm第二球形颗粒,约140kg,小于1.7mm的球形颗粒倒回转盘成型机中继续长大。
[0147]
将上述得到的90kg第二球形颗粒在45℃下吹风,中途向滚动成型机多次补充微量水,收紧2小时,在120℃干燥24小时,之后在550℃下焙烧10小时。最终得到分子筛含量100%的焙烧产物。
[0148]
(5)将90kg上述焙烧产物与900kg碱性缓冲溶液(该碱性缓冲溶液为氨水和硝酸铵水溶液的混合液,其中,氨水的含量为26重量%,硝酸铵水溶液中硝酸铵的含量为7.5重量%,氨水与硝酸铵水溶液的重量比为3:2,碱性缓冲溶液的ph值为11.35)加入到带压反应釜中,在80℃、2.3kg/cm2压力下搅拌1小时,然后洗涤、过滤、干燥,得到催化剂编号为a4。
[0149]
测得催化剂a4的压碎强度σ列于表1。
[0150]
实施例5
[0151]
(1)在25℃恒温下分别将384kg甲醇、87.5kg三正丙胺和180kg22.5%的四丙基氢氧化铵水溶液加入到3m3不锈钢反应釜中,补加900kg水和17.6g zrocl2·
8h2o倒入反应釜中,搅拌10分钟,分四批次、每批次间隔15分钟,将304.6kg正硅酸甲酯加入上述混合溶液中,搅拌60分钟,之后在25℃恒温下继续搅拌水解2小时,形成ph值为12.44的胶体混合物,其中,硅源:(c3h8)3n:tpaoh:meoh:h2o的用量摩尔比=1:0.30:0.10:6:30,硅源与zr
4+
的用量质量比为23600:1,其中,硅源以sio2计。
[0152]
(2)将上述胶体混合物先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天。
[0153]
(3)将反应釜降温,打开釜盖,在75℃温度下进行6小时的赶甲醇处理,然后采用50nm六管膜进行膜过滤并采用50℃的水进行洗涤,至洗涤水ph值达到或接近9.0,洗涤后得到的分子筛浆液在120℃干燥20小时,得到本实施例滚动成型所需的分子筛原粉。
[0154]
取少量上述得到的分子筛原粉在550℃焙烧6小时,得到的分子筛中,锆离子含量为41μg/g。该分子筛晶粒大小均匀,粒径为0.15-0.25μm。
[0155]
(4)将步骤(3)得到的分子筛原粉在粉碎机中粉碎,取2kg200-500目的粉碎后分子筛原料置于转盘成型机中,所用转盘滚动成型机的转盘直径1.2m,转盘深度为450mm,转盘
倾角确定为50
°
,转盘转速设定30rpm。向其中喷洒去离子水1.5kg,得到直径大约0.2-0.8mm的第一球形颗粒。
[0156]
另取220kg 200-800目的粉碎后分子筛原料与100kg的sio2含量为30重量%的碱性硅溶胶混合均匀并重新粉碎,取用小于30目的颗粒向上述具有第一球形颗粒的转盘成型机中匀速加入300kg,240min内加完。其中,中途多次用12目和9目的筛子筛分,得到直径1.7-2.2mm第二球形颗粒,约160kg,小于1.7mm的球形颗粒倒回转盘成型机中继续长大。
[0157]
将上述得到的90kg第二球形颗粒在45℃下吹风,中途向滚动成型机多次补充微量水,收紧2小时,在120℃干燥24小时,之后在530℃下焙烧10小时。最终得到分子筛含量86%的焙烧产物。
[0158]
(5)将90kg上述焙烧产物与900kg碱性缓冲溶液(该碱性缓冲溶液为氨水和硝酸铵水溶液的混合液,其中,氨水的含量为26重量%,硝酸铵水溶液中硝酸铵的含量为7.5重量%,氨水与硝酸铵水溶液的重量比为3:2,碱性缓冲溶液的ph值为11.35)加入到带压反应釜中,在80℃、2.3kg/cm2压力下搅拌1小时,然后洗涤、过滤、干燥,得到催化剂编号为a5。
[0159]
测得催化剂a5的压碎强度σ列于表1。
[0160]
实施例6
[0161]
(1)在25℃恒温下分别将575kg甲醇、43.75kg三正丙胺和270kg22.5%的四丙基氢氧化铵水溶液加入到3m3不锈钢反应釜中,补加510kg水和13.2g h2ptcl6·
6h2o倒入反应釜中,搅拌10分钟,分四批次、每批次间隔15分钟,将304.4kg正硅酸甲酯加入上述混合溶液中,搅拌60分钟,之后在25℃恒温下继续搅拌水解2小时,形成溶胶,搅拌均匀,形成ph值为12.82的胶体混合物,其中,硅源:(c3h8)3n:tpaoh:meoh:h2o的用量摩尔比=1:0.15:0.15:9:20,硅源与pt
4+
的用量质量比为23600:1,其中,硅源以sio2计。
[0162]
(2)将上述胶体混合物先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天。
[0163]
(3)将反应釜降温,打开釜盖,在75℃温度下进行6小时的赶甲醇处理,然后采用50nm六管膜进行膜过滤并采用50℃的水进行洗涤,至洗涤水ph值达到或接近9.0,洗涤后得到的分子筛浆液在120℃干燥20小时,得到本实施例滚动成型所需的分子筛原粉。
[0164]
取少量上述得到的分子筛原粉在550℃焙烧6小时,得到的分子筛中铂离子含量为41μg/g。该分子筛晶粒大小均匀,粒径为0.15-0.25μm。
[0165]
(4)将步骤(3)得到的分子筛原粉在粉碎机中粉碎,取2kg100-1000目的粉碎后分子筛原料置于转盘成型机中,所用转盘成型机的转盘直径1.2m,转盘深度为450mm,转盘倾角确定为50
°
,转盘转速设定30rpm。向其中喷洒去离子水1.5kg,得到直径大约0.2-0.8mm的第一球形颗粒。
[0166]
另取220kg 200-800目的粉碎后分子筛原料与100kg的sio2含量为30重量%的碱性硅溶胶混合均匀并重新粉碎,取用小于30目的颗粒向上述具有第一球形颗粒的转盘成型机中匀速加入300kg,240min内加完。其中,中途多次用12目和9目的筛子筛分,得到直径1.7-2.2mm第二球形颗粒,约160kg,小于1.7mm的球形颗粒倒回转盘成型机中继续长大。
[0167]
将上述得到的90kg第二球形颗粒在45℃下吹风,中途向滚动成型机多次补充微量水,收紧2小时,在120℃干燥24小时,之后在550℃下焙烧10小时。最终得到分子筛含量86%的焙烧产物。
[0168]
(5)将90kg上述焙烧产物与900kg碱性缓冲溶液(该碱性缓冲溶液为氨水和硝酸铵水溶液的混合液,其中,氨水的含量为26重量%,硝酸铵水溶液中硝酸铵的含量为7.5重量%,氨水与硝酸铵水溶液的重量比为3:2,碱性缓冲溶液的ph值为11.35)加入到带压反应釜中,在100℃、2.8kg/cm2压力下搅拌1小时,然后洗涤、过滤、干燥,得到催化剂编号为a6。
[0169]
测得催化剂a6的压碎强度σ列于表1。
[0170]
实施例7
[0171]
按照实施例1的方法,不同的是,将三正丙胺替换为相同摩尔量的乙二胺。得到催化剂编号a7。
[0172]
测得催化剂a7的压碎强度列于表1。
[0173]
实施例8
[0174]
按照实施例3的方法,不同的是,将ce(no3)4·
7h2o替换为zrocl2·
8h2o,且硅源与zr
4+
的用量质量比为40000:1,得到的分子筛中,锆离子含量为24.2μg/g。得到催化剂编号为a8。测得催化剂a8的压碎强度列于表1。
[0175]
实施例9
[0176]
按照实施例3的方法,不同的是,将ce(no3)4·
7h2o替换为铝源(sb粉,氧化铝质量含量为70%,ti
4+
离子含量5μg/g),且硅源与al
3+
的用量质量比为15000:1,得到的分子筛中,铝离子含量为64.1μg/g。得到催化剂编号为a9。测得催化剂a9的压碎强度列于表1。
[0177]
实施例10
[0178]
按照实施例1的方法,不同的是,步骤(4)包括:将步骤(3)得到的分子筛原粉在粉碎机中粉碎,取220kg 200-800目的粉碎后分子筛原料与100kg的sio2含量为30重量%的碱性硅溶胶置于转盘成型机中,所用转盘成型机的转盘直径1.2m,转盘深度为450mm,转盘倾角确定为50
°
,转盘转速设定30rpm。用12目和9目的筛子过筛,得到直径1.7-2.2mm第二球形颗粒。得到催化剂编号为a10。测得催化剂a10的压碎强度列于表1。
[0179]
试验例1
[0180]
本试验例1用于说明实施例1-10和对比例1-5所制备的分子筛催化剂在气相贝克曼重排反应中的催化反应结果。
[0181]
分别采用催化剂a1-a10和d1-d5在试验条件1和试验条件2上进行环己酮肟气相贝克曼重排反应。
[0182]
试验条件1:反应装置为常压连续流动固定床,反应器内径为5毫米,催化剂的装填量0.469克,催化剂床层上面装填约30mm高30目的粗石英砂,催化剂床层下面装填50目的细石英砂。催化剂粒度20-60目。催化剂在装入反应管后,在常压、350℃的氮气气氛中预处理1小时。原料环己酮肟的浓度为35重量%,重量空速(whsv)为16h-1
,溶剂为甲醇,反应温度为380℃,氮气流量为45ml/min,反应产物经冰水混合物冷却后进入收集瓶进行气液分离,反应时间6小时进行产物组成分析。
[0183]
试验条件2:反应装置为连续流动固定床,反应器内径为28毫米,反应压力:0.1mpa;反应温度:380℃;n2:环己酮肟(摩尔比)=12:1;水在环己酮肟的甲醇溶液中的质量百分比为1.2wt%;汽化器控温175℃;管线保温185℃;催化剂的装填30g;床层高度:15.0cm;原料环己酮肟的浓度为35重量%,重量空速(whsv)0.5h-1
,溶剂为无水甲醇,反应时间600小时进行产物组成分析。
[0184]
反应产物采用agilent公司6890型气相色谱仪(氢焰离子检测器,peg20m毛细管色谱柱,柱长50m)进行定量分析,汽化室温度250℃,检测室温度为240℃,柱温为程序升温,110℃恒温8分钟,15℃/min升到230℃再恒温14分钟。反应结果见表1。
[0185]
反应后己内酰胺和环己烯酮的重排产物含量采用面积归一法计算,溶剂不参与积分。
[0186]
通过上述分析得到反应产物中环己酮肟摩尔百分含量以及反应产物中己内酰胺摩尔百分含量,根据下述公式计算出环己酮肟转化率和己内酰胺总选择性。结果如表1所示。
[0187]
环己酮肟转化率(mol%)=(100-反应产物中环己酮肟摩尔百分含量)/100
×
100%
[0188]
己内酰胺总选择性(mol%)=反应产物中己内酰胺摩尔百分含量/(100-反应产物中环己酮肟摩尔百分含量)
×
100%
[0189]
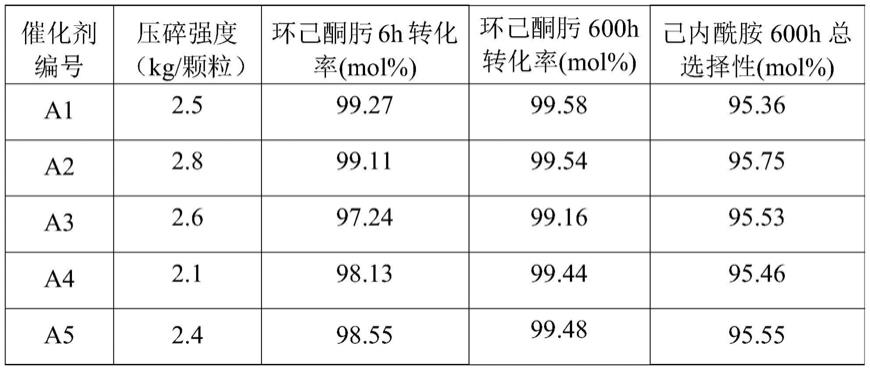
表1
[0190][0191]
[0192]
从表1可以看出,本发明的方法制得的分子筛催化剂具有较高的压碎强度,最高可达到2.9kg/颗粒,因此可以用于环己酮肟气相贝克曼重排制备己内酰胺的固定床或移动床工艺。另外,本发明制得的催化剂a1-a10的环己酮肟转化率较高,反应6小时后环己酮肟的转化率可以达到97%以上,与现有技术cn1338427a中方法所合成的全硅分子筛催化剂相比,环己酮肟的转化率提高了1%。另外,实例1-10合成的催化剂用于反应600小时后,环己酮肟的转化率仍可达到99%以上,催化剂的寿命较长。在相同的试验条件下对比例1-5的全硅分子筛作催化剂用于环己酮肟气相贝克曼重排反应进行600小时后催化剂失活,环己酮肟转化率明显低于本发明提供的催化剂。
[0193]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有1条留言
-
 0访客 来自[中国] 2022年11月15日 19:45解释的特别清楚,满分
0访客 来自[中国] 2022年11月15日 19:45解释的特别清楚,满分
1