金属碳化物纳米材料催化剂及其制备方法与流程
相关申请的交叉引用
本申请要求申请号62/800,662,2019年2月4日提交的标题为“金属碳化物纳米材料及其制备方法和催化应用”的美国临时申请,以及申请号16/744,383,2020年1月16日提交的标题为“金属碳化物纳米材料催化剂及其制备方法”的美国申请的权益,其通过引用结合在本申请中。
本发明涉及碳氢化合物转化催化剂及其生产方法,更具体地,涉及新型纳米材料催化剂的合成,该新型纳米材料催化剂包括封装的金属碳化物纳米团簇,其中该金属碳化物纳米团簇是在相对温和的条件、例如100-700℃的温度和大气条件下,通过用碳氢化合物处理限制的金属纳米团簇而制备的。
本发明还涉及使用新型金属碳化物纳米材料的用于碳氢化合物转化的催化工艺,其中催化材料包括限制的金属碳化物纳米团簇,其对碳氢化合物转化是高活性的,其中金属碳化物中的金属为过渡金属。
背景技术:
近年来,基于金属碳化物的催化材料因其在许多反应中的独特催化性能而备受关注。例如,《科学》320(2008)86-89报道了一种钯(pd)碳化物催化剂的催化材料可以催化炔烃氢化选择性。催化剂中碳的存在对控制产物的选择性起着关键作用。《催化科学与技术》7(2017)807-816证明了合成铂(pt)碳化物纳米材料是可行的,其中该材料表现出独特的催化性能。然而,实际上,pt碳化物材料只有在例如高温高压的相当极端的条件下曾经成功合成过。例如,《固态通信》133(2005)55-59教导了,pt碳化物材料可以通过激光加热的金刚石对顶砧(diamondanvilcell)上负载的pt粉的高压、高温处理来合成。高压x射线衍射实验证实了,在85gpa和2600k的合成条件下,碳化物结构可以通过pt与碳载体的相互作用而形成。
然而,通过高压和高温处理制备的pt碳化物材料不仅昂贵,而且应用非常有限。因此,有必要开发一种可以在相对温和的条件下制备pt碳化物材料的新的合成方法。
美国页岩气的繁荣也为开发用于碳氢化合物转化的先进催化剂和工艺提供了机会,例如,如在通过引用并入本文中的美国专利7,153,807和8,946,107以及美国公开号2018/0194701中所教导的。实际上,大多数催化工艺都基于非均相催化剂。与均相催化材料相比,非均相催化材料更容易制备、运输和处理,而且往往更稳定、制造成本更低。
例如,诸如乙烯、丙烯和丁烯的低碳烯烃广泛应用于聚合工业。这些烯烃产品通常含有不饱和炔烃和烯烃杂质,如乙炔和丁二烯,它们是通过蒸汽裂解或流化催化裂解产生的副产品。这些杂质的存在通常会抑制聚合催化材料的活性,因此必须清除。一般来说,炔烃和二烯烃选择性加氢成烯烃是工业装置最有吸引力的解决方案。因此,钯基催化剂在这种加氢反应中得到了大量的研究和广泛的应用。
然而,目前商业化的钯基催化剂存在的问题是产生大量的饱和物和绿油(c4+低聚物化合物)副产物,这些副产物是由于烯烃的过度加氢和/或炔烃和/或二烯烃和/或烯烃的齐聚反应而产生的。这种绿油副产物由于其对烯烃获得选择性的不利影响而是不希望的。更重要的是,绿油还会在加氢催化剂表面上沉积c4+化合物,从而抑制催化剂的活性。也就是说,绿油降低了加氢催化剂的寿命,例如,使催化剂无法有效地辅助碳氢化合物反应。因此,非常期望开发新型催化剂,其可以选择性加氢炔烃和二烯烃成具有降低的绿油产率的烯烃。
美国专利7,153,807公开了pt基ptir和pt基ptru双金属催化剂以及pt基ptruag三元催化剂,以用于炔烃和二烯烃选择性加氢成具有低绿油选择性的烯烃。美国专利7,153,807还描述了使用负载在二氧化硅或氧化铝载体上的ptir和ptru双金属催化剂以及ptruag三元催化剂,以将炔烃和二烯烃选择性加氢成烯烃的工艺。与商业pd基催化剂相比,这些pt基催化剂对乙炔加氢表现出高活性,降低的绿油选择性。此外,这些pt基催化剂的额外优点还包括由于绿油产率的抑制而延长催化剂的使用寿命或延长操作周期,从而降低了催化剂的抑制。
美国公开号2004/0176652还教导了负载在氧化铝上的pt基ptru、pt基ptruag和pt基ptruga催化剂,这些催化剂用于双床工艺(dualbedprocess)中以选择性地将炔烃和二烯烃加氢成烯烃。美国专利9,533,288描述了一种pt基金属催化剂的制备,其中ni层电镀在金属载体上,该金属载体随后具有电镀的pt的顶层。这种pt基负载型金属催化剂对不饱和碳氢化合物的选择性加氢表现出高的活性。
表1示出了在美国专利7,153,807中报告的这种pt基催化剂的测试结果。与商业pd/ag基催化剂相比,al2o3上0.6%pt示出了较高的乙烯选择性和略低的绿油选择性,但更低的乙炔转化率。虽然ir本身不活跃,但通过向pt中加入ir,可以显著提高乙炔转化率,并进一步降低绿油的选择性。然而,在这种情况下,乙烯选择性大大降低,这是ptir双金属催化剂的一个显著缺点。对ptru和ptruag催化剂也观察到类似的性能。总的来说,pt/al2o3催化剂表现出高的乙烯选择性,但是低的乙炔转化率,和高的绿油产率;而pt基双金属和pt基三金属催化剂表现出改善的乙炔转化率,降低的绿油产率,但是更低的乙烯选择性。
然而,好的加氢催化剂必须能够在保持高活性和高乙烯选择性的情况下,最小化绿油的产率。这样一来,无论是美国专利7,153,807中所述的pt基双金属和pt基三金属催化剂,还是美国公开号2004/0176652和美国专利9,533,288中所公开的催化剂都不符合该标准。
表1
此外,乙烷直接转化为芳族化合物作为从廉价且丰富的来源生产轻芳族化合物(苯、甲苯、二甲苯或苯系物(btx))的替代方法是非常令人感兴趣的。迄今为止,贵金属改性的沸石催化剂在乙烷芳构化反应中得到了广泛的研究,其中这些催化剂通常含有0.02至0.5wt%的负载有贵金属的金属。
《催化科学与技术》8(2018)1500-1516对该领域的产业参与者的早期专利以及科学论文提供了全面的探索,特别是,乙烷直接转化为芳族化合物的催化剂的制备及其催化性能。此外,美国专利8,946,107和美国公开号2018/0194701公开了基于pt改性的zsm-5催化剂将乙烷选择性转化为芳族化合物的工艺。
例如,美国专利8,946,107公开了一种使用具有pt负载量为0.005至0.1wt%的pt/zsm-5催化乙烷芳构化反应的方法。美国专利8,946,107还描述了向pt中添加诸如fe的第二种金属,可以降低不期望的甲烷产率。但这种双金属催化剂也降低了催化剂在乙烷转化率方面的活性。
虽然有报道称,pt/zsm-5基催化剂对乙烷芳构化具有活性,但《催化科学与技术》8(2018)1500-1516、美国专利8,946,107和美国公开号2018/0194701以及其他文献中并未提及乙烷芳构化过程中pt的活性相和化学状态。换句话说,所有的这类催化剂的早期研究都未能提供关于乙烷芳构化反应中的pt/zsm-5基催化剂的催化剂配方和活性相的完整信息。因此,了解该类催化剂的催化剂配方和活性相的最佳条件是有益的。
技术实现要素:
本发明通过以下几种方法提供对现有技术方法和催化剂的改进来解决现有技术的缺陷。例如,本发明提供了可以在其他不饱和化合物存在下选择性加氢炔烃和二烯烃的催化剂,制备该催化剂的方法以及在催化应用中使用该催化剂的方法。
为了实现本发明的目的,本发明的第一方面涉及催化剂,其包括可通过各种技术表征的新型的非均相金属碳化物纳米材料。本发明还涉及一种新型制备方法,以在相对温和的条件下合成金属碳化物纳米材料,以在载体和/或粘结剂中形成封装的贵金属和/或贵金属碳化物纳米团簇,其中,以前在任何可用的文献中从未报道过在温和条件下形成这些金属碳化物纳米材料。例如,催化剂包括限制的铂碳化物纳米团簇,其通过在高温下用乙烷处理封装的铂纳米团簇而制备。
本发明的第二方面涉及将这种新型的非均相金属碳化物纳米材料催化材料应用于催化应用,该新型的非均相金属碳化物纳米材料催化材料包括在载体和/或粘结剂上限制或封装的金属碳化物纳米团簇,该金属碳化物纳米团簇对选择性碳氢化合物转化为高活性的,其中所述金属优选地为贵金属。
在实现本发明的这些和其他目的时,金属碳化物纳米材料催化剂可包含一种或多种过渡金属。过渡金属的示例包括但不限于铂、铜、镍、铼、钽、锰、铱和锇;优选地为铂。过渡金属负载在至少一种载体和/或粘结剂上,所述载体和/或粘结剂选自于、但不限于无机氧化物、硅碳化物、硅氮化物、硼氮化物、碳及其组合,其中优选地,所述载体为铝硅酸盐沸石。沸石载体的示例包括但不限于zsm-5(zeolitesoconymobil-5)、zsm-11、zsm-12、zsm-23、zsm-35、丝光沸石(mordenite,mor)、镁碱沸石、八面沸石、白沸石、β沸石(bea)、y沸石、x沸石、ssz-13沸石、钛硅分子筛-1(ts-1)、斜碱沸石、板沸石、斜发沸石、交沸石、浊沸石、方碱沸石、铯沸石和介孔二氧化硅(例如,mcm41)。
金属碳化物纳米材料催化剂可用于催化选择性碳氢化合物转化。例如,发现含有限制的/封装的金属碳化物纳米团簇的催化剂可以在近室温下有效地、选择性地催化炔烃和二烯烃加氢成烯烃,具有较高烯烃选择性和可忽略不计的绿油产率。此外,还发现包括限制的金属碳化物纳米团簇的催化剂对乙烷直接转化为芳族化合物显示出高的活性。
使用本发明包括以下几个益处:
1.只生产微量的绿油,从而显著地提高了催化剂的使用寿命。
2.在相同的反应条件下,在保持相同的乙烯选择性的情况下,比标准pt/al2o3催化剂表现出更高的催化活性。
附图说明
本文中的附图用于提供对本发明的进一步理解,并构成说明书的一部分。附图与以下实施方案一起用于解释本发明,但不构成对本发明的限制。在附图中:
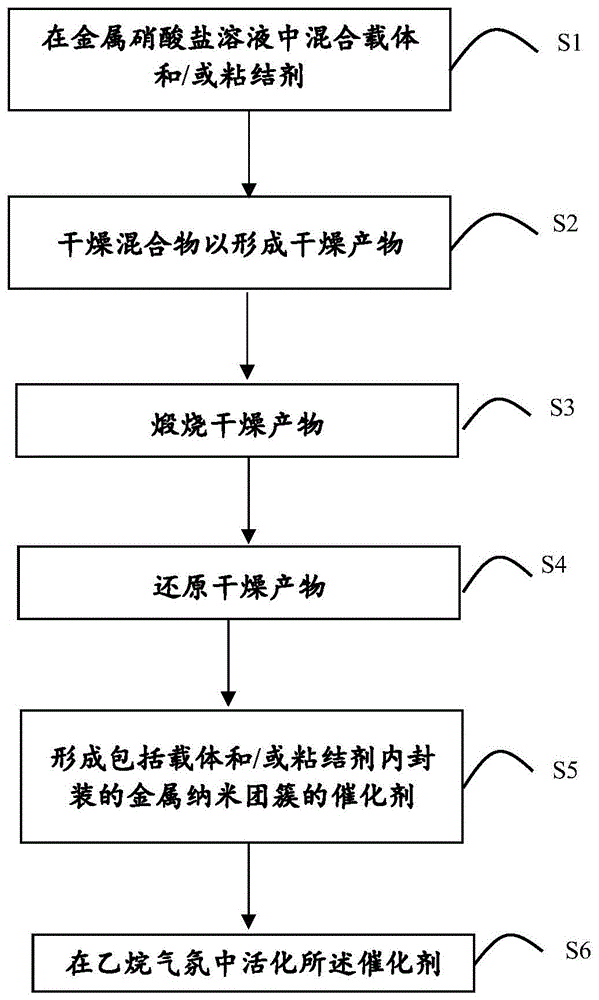
图1是形成金属纳米材料催化剂的过程的示意图。
图2是现有技术催化剂(还原后的pt/al2o3)和本发明第一方面体现的催化剂在630℃原位氢还原后(还原后的pt/zsm-5)的动态co化学吸附数据的对比图。
图3是现有技术催化剂(还原后的pt/al2o3)、本发明第一方面体现的催化剂(还原后的pt/zsm-5)、本发明第二方面体现的催化剂(乙烷活化后的pt/zsm-5)和乙烷活化后的现有技术催化剂(乙烷处理后的pt/al2o3)的co-drift光谱的对比图。
图4是示出本发明第一方面体现的催化剂(还原后的pt/zsm-5)和本发明第二方面体现的催化剂(乙烷活化后的pt/zsm-5)的ftir光谱的视图。
图5是示出本发明第一方面体现的催化剂(还原后的pt/zsm-5)和本发明第二方面体现的催化剂(乙烷活化后的pt/zsm-5)以及作为参考的pt箔的原位xanes光谱的视图。
图6是示出在选择性乙炔加氢反应中,现有技术催化剂(还原后的pt/al2o3)、体现本发明第一方面的催化剂(还原后的pt/zsm-5)和体现本发明第二方面的催化剂(乙烷活化后的pt/zsm-5)在乙炔转化方面的催化性能的视图。
图7是示出在选择性乙炔加氢反应中,现有技术催化剂(还原后的pt/al2o3)、体现本发明第一方面的催化剂(还原后的pt/zsm-5)和体现本发明第二方面的催化剂(乙烷活化后的pt/zsm-5)的绿油选择性的视图。
图8是示出了在250-620℃温度范围内,体现本发明第一方面的催化剂(还原后的pt/zsm-5)上乙烷的程序升温表面反应数据的视图。
图9是示出了在250-620℃温度范围内,体现本发明第二方面的催化剂(乙烷活化后的pt/zsm-5)上乙烷的程序升温表面反应数据的视图。
图10是显示了体现本发明第一方面的催化剂(还原后的pt/zsm-5)和体现本发明第二方面的催化剂(乙烷活化后的pt/zsm-5)上甲烷和btx产率作为反应温度的函数的视图。
在各个附图中,相似的元件提供有相似的附图标记。应注意的是,附图不必须按比例或占比绘制,而是为了提供对其组件的更好理解而绘制,并且不旨在限制范围,而是提供示例性说明,并且绘制图形是为了呈现相关数据。
具体实施方式
下面将参照附图详细描述本发明的实施方式。应当理解,本文所描述的实施例仅用于本发明的说明和解释,而不用于限制本发明。
本发明涉及一种合成负载型催化剂的方法,其中活性相位于固体载体的表面或孔内,其中新型负载型催化剂能够实现具有高烯烃获得选择性和低绿油(低聚物)和/或饱和物选择性的选择性加氢性能,并且涉及由此方法获得的催化剂。这样,本发明至少具有增加催化剂预期寿命和/或增加催化活性的优点。
如图1中所示,在本发明的一个实施方案中,提供了一种合成新型非均相催化剂的方法,该方法至少包括以下步骤中的一些:
步骤1(s1):在金属前体、优选金属硝酸盐的溶液中混合载体和/或粘结剂,以形成混合物;
步骤2(s2):干燥混合物,以形成干燥产物;
步骤3(s3):煅烧干燥产物;
步骤4(s4):还原干燥产物;
步骤5(s5):形成包括载体和/或粘结剂内封装的金属纳米团簇的催化剂;和
步骤6(s6):优选地,但也不必须地,在乙烷气氛中的活化催化剂。
具体地,在混合步骤s1中,载体和/或粘结剂选自无机氧化物、硅碳化物、硅氮化物、硼氮化物、碳、沸石及其组合,其中优选地,所述载体是铝硅酸盐沸石,其包括但不限于zsm-5、zsm-11、zsm-12、zsm-23、zsm-35、mor、镁碱沸石、八面沸石、菱沸石、β沸石(bea)、y沸石、x沸石、ssz-13、钛硅分子筛(ts-1)、斜碱沸石、板沸石、斜发沸石、交沸石、浊沸石、方碱沸石、铯沸石和介孔二氧化硅(例如mcm41),优选为zsm-5,具有硅铝比10:1到80:1,优选地20:1到45:1,最优选地在30:1。将载体和/或粘结剂与水溶液混合,该水溶液包含金属前体、优选硝酸铂,该硝酸铂包括铂盐和铂络合物,其中该混合在例如10-100℃、优选80℃的温和温度和例如标准大气压101.3kpa的环境压力下混合。
然后在步骤s2干燥混合物以获得干燥产物,其中干燥产物随后在步骤s3在例如在300-800℃、优选地在550℃的高温下空气煅烧一定时间、优选地0.5到24小时,更优选地4小时。值得注意的是,在空气煅烧步骤s3期间,在空气煅烧步骤之前或之后可以添加惰性粘结剂,例如二氧化硅粉末和/或氧化铝粉末,以提高催化剂强度。空气煅烧后,产物在步骤s4中,在例如300-800℃、优选630℃的高温下在氢气中原位还原一定时间,优选地0.5到24小时,更优选地1小时,然后在氢气气氛中冷却至室温,例如20-25℃,以在步骤s5中提供合成的催化剂。
任选地,在原位还原之后,在步骤s6,产物在氢气气氛中冷却至100-650℃,优选地300-500℃,更优选地400℃。随后在300-500℃,优选地400℃温度下用诸如氮气的惰性气体清洗,并在包含至少一个含碳分子和氮气(例如,50%的氮平衡)的气氛中,在100-750℃、优选地300-750℃、更优选地400℃的温度下活化一定时间,该至少一个含碳分子例如为cox、csx、cxhy、cxhyclz、cxhyfz、cxhybriz、cxhyiz、cxhyoz,优选地为乙烷。活化后,催化剂使用含碳分子气氛冷却至80-150℃之间、优选至100℃,然后在惰性气氛、例如氮气中进一步冷却至室温。
所得加氢催化剂是一种非均相催化剂,其包括载体和/或粘结剂上的金属纳米团簇,以及更优选地铝硅酸盐沸石上的铂或铂碳化物纳米团簇,其中金属或金属碳化物纳米团簇限制或封装在沸石的微孔中。例如,金属或金属碳化物纳米团簇具有接近1nm、例如+/-10%的尺寸,以封装在沸石中。在本发明的一个实施方案中,催化剂包含300-25000ppm的铂,且最优选为500ppm的铂,其中该铂具有大于90%、且优选地95-100%的金属分散性。
本发明方法和催化剂将进一步使用具体实施例来说明,这些具体实施例仅用于说明本发明,但不以任何方式限制本发明。
比较实施例1
将5克al2o3粉末与一定量的pt(no3)2溶液混合。将混合物在室温下搅拌1小时,然后在旋转蒸发器中加热至80℃1小时,以获得干燥产物,然后在550℃下空气煅烧4小时。然后,将粉末在630℃的氢气中原位还原1小时,并在氢气气氛中冷却至室温。然后将粉末压制并筛分至20x40目。所得产物表示为催化剂a,其含有500ppmpt。然后对催化剂a进行表征测量以确定其特征。
实施例1
将5g的zsm-5粉末(具有硅铝比为30)与一定量的pt(no3)2在溶液中混合。将混合物在室温下搅拌1小时,然后在旋转蒸发器中加热至80℃以获得干燥产物,随后在550℃下空气煅烧4小时。然后,粉末在630℃,在氢气中原位还原1小时,并在氢气气氛中冷却至室温(例如,在20-25℃之间)。然后将粉末压制并筛分至20x40目。所得产物表示为催化剂b,其含有500ppmpt。然后对催化剂b进行表征测量以确定其特征。
实施例2
使用与实施例1中催化剂b相同的步骤制备粉末,但在氢气气氛中还原后,粉末在氢气气氛中冷却至400℃,随后在400℃下进行惰性气体清洗10分钟,然后在300-500℃的温度下在乙烷(50%氮平衡)中活化。乙烷活化后,粉末在乙烷流下冷却至100℃,并在惰性气体气氛、例如氮气中进一步冷却至室温。所得产物表示为催化剂c,其还含有500ppmpt。
然后通过动态co化学吸附测量(化学吸附)通过测试和分析催化剂中催化剂的铂分散度来对催化剂a、b和c进行表征,其中,通过漫反射红外傅里叶变换光谱(drifts,diffusereflectanceinfraredfouriertransformspectroscopy)和x射线吸收近边结构(xanes,x-rayabsorptionnearedgestructure)研究铂类的演变。如下文将述,催化剂b的表征数据显示,在还原后,铂形成封装的pt小纳米团簇。此外,在乙烷活化后(催化剂c),这些pt纳米团簇转化为新型封装的pt碳化物纳米团簇ptcx/zsm-5。数据还表明,添加惰性粘结剂不会影响这种转化,换句话说,不会影响从pt纳米团簇到pt碳化物纳米团簇的形成。
图2示出了在630℃下原位氢还原后的催化剂a(曲线200)和在630℃下原位氢还原后的催化剂b的动态co化学吸附数据(曲线220)。动态co化学吸附确定的pt分散度和平均粒径在表2中列出。图2和表2表明,对于催化剂a,pt/al2o3,氢还原后,pt分散度约为21%,pt主要形成平均粒径为6nm的纳米颗粒。另一方面,对于还原后的催化剂b,数据表明pt的分散度约为100%,铂形成团簇尺寸接近1nm的小的纳米团簇。此外,数据表明催化剂b具有更大的峰宽,这表明这些纳米团簇位于沸石的微孔内。换句话说,数据表明,还原后,催化剂b包括沸石载体中封装的铂纳米团簇。
图3示出了还原后的催化剂a(还原后的pt/al2o3)(曲线300),还原后的催化剂b(还原后的pt/zsm-5)(曲线320)和催化剂c(乙烷活化后的pt/zsm-5)(曲线340),以及乙烷处理后催化剂a的示例(“改性的催化剂a”)(曲线360)的co-drift光谱。在催化剂a和催化剂b的情况,样品首先在630℃下用氢还原,然后在氦气气氛中冷却至室温,随后在室温下进行co-drifts测量。在催化剂c和改性的催化剂a的情况,在相同的处理后,催化剂在乙烷中冷却至100℃,随后在氦气中进一步冷却至室温。在额外的氦气清洗30分钟后,在室温下进行co-drifts测量。
图3中的co-drifts测量表明,对于还原后的催化剂a,pt形成金属纳米粒子,这与图2中的动态co化学吸附测量一致。然而,对于还原后的催化剂b,co-drifts和动态co化学吸附数据表明,大部分铂形成了沸石内封装的微孔内的pt小纳米团簇。此外,co-drift光谱还表明,乙烷活化后,与pt纳米团簇相关的峰完全消失,而在2250cm-1-2150cm-1范围内出现了新的双峰特征。类似的双峰特征在钼碳化物材料的co-drifts研究(《acs应用材料与界面》,9(2017)9815-9822)中也报道了。因此,可以理解的是,结合下面列出的其他特征,这种双峰特征可能与铂碳化物纳米团簇的存在有关。因此,可以认为,还原后pt/zsm-5催化剂中存在的封装的pt纳米团簇在乙烷活化后转变为催化剂c、pt/zsm-5中封装的pt碳化物纳米团簇。另一方面,图3还示出,对于类似乙烷处理后的催化剂a,不存在如催化剂c的双峰特征,这表明在乙烷处理时,大的pt金属纳米粒子不能转化为铂碳化物种类。
图4示出了还原后的催化剂b(还原后的pt/zsm-5)(曲线420)和催化剂c(乙烷活化后的pt/zsm-5)(曲线440)的ftir光谱。在催化剂b的情况,还原后,样品在氦气中冷却至室温,然后在室温下进行ftir测量。在催化剂c的情况,乙烷活化后,样品首先在乙烷中冷却至100℃,然后在氦气中进一步冷却至室温,随后在ftir测量之前在室温下氦气清洗30分钟。
如图4中所示,催化剂b在pt-c拉伸范围内的ftir光谱清楚地示出,还原后,没有与pt-c键相关的峰。然而,在乙烷活化后,如文献(《化学物理快报》,560(2013)42-48)中所述,对于催化剂c有多个与pt-c键相关的峰。因此,原位ftir数据表明,乙烷活化后,催化剂c中存在铂碳化物类。
图5示出了还原后的催化剂b(还原后的pt/zsm-5)(曲线520)和催化剂c(乙烷活化后的pt/zsm-5)(曲线540)以及作为参考的pt箔(曲线580)的原位xanes光谱。在催化剂b的情况,还原后,样品在氦气中冷却至室温,然后在室温下进行xanes测量。在催化剂c的情况,乙烷活化后,样品首先在乙烷中冷却至100℃,然后在氦气中进一步冷却至室温,随后在xanes测量之前在室温下氦气清洗30分钟。
图5中的xanes数据进一步支持在乙烷活化后形成了封装的pt碳化物纳米团簇。例如,在还原后,催化剂b的xanes光谱不同于pt金属箔,其中在还原后的pt/zsm-5的情况下存在高强度白线。类似地,在mcm-22微孔中封装的典型pt小纳米团簇的已公开的研究(《自然材料》,16(2017)132-138)中观察到了这种具有高强度白线的xanes光谱。因此,xanes光谱还表明,在还原后,pt在催化剂b中形成小的纳米团簇。此外,在乙烷活化后,催化剂c的xanes光谱中出现了11530-11560ev之间的新的前边缘峰。pt的类似的前边缘峰在碳基质中嵌入的原子分散的pt的已公开的研究(《科学进展》,4(2018)eaao6657)中已被报道,且归因于限制的pt类的pt-c键的存在对pt的电子效应。催化剂c的前边缘峰的出现很可能也是由于pt-c键的形成对pt的电子效应所致。因此,xanes数据也支持了乙烷活化后,pt纳米团簇向pt碳化物纳米团簇转化的结论。
也就是说,这些催化材料的特性如图2至图5和表2(如下)所示。
表2
发现这种新型催化剂在本领域已知的氢转化工艺期间能够实现改进的选择性加氢化性能,其具有高的烯烃获得选择性和低的绿油和/或饱和物选择性。例如,如美国专利7,153,807和8,946,107以及美国公开号2018/0194701中公开的氢转化工艺,其通过引用并入本文中。这样,新型催化剂b和c的优点包括但不限于由于绿油产量的降低而延长催化剂的使用寿命和/或延长加氢操作周期。
在本发明的一个实施方案中,乙烷芳构化在300-750℃、优选地500-650℃、更优选地600-630℃的温度下和/或在500-5000hr-1的乙烷气体时空速度(ghsv,gashourlyspacevelocity)、优选1000hr-1的乙烷ghsv下进行。此外,乙炔选择性加氢成乙烯在20-200℃、优选地20-90℃的温度下和/或在10-1000hr-1的乙炔ghsv、以及0.5-20、优选地2-6的h2/c2h2比率下进行。
下面将关于如在选择性乙炔加氢反应和乙烷芳构化反应、例如炔烃和二烯烃催化加氢成烯烃和乙烷催化转化成芳族化合物中示出的碳氢化合物转化中的催化剂a、b和c的催化性能,进一步讨论这种新型催化剂b和c的优点。申请人注意到,虽然讨论了将乙炔选择性加氢成乙烯的催化碳氢化合物转化工艺,但这种转化工艺不限于所述的将乙炔选择性加氢成乙烯,还可以包括1,3-丁二烯(c4h6)选择性加氢成丁烯(c4h8)。这些催化剂中的pt分散度通过动态co化学吸附测量来检测。这些催化剂的活性相通过co-drifts和xanes来研究。
图6示出了在选择性乙炔加氢反应中催化剂a(还原后的pt/al2o3)(曲线700)、催化剂b(还原后的pt/zsm-5)(曲线720)和催化剂c(活化后的pt/zsm-5)(曲线740)的乙炔转化率的催化性能。如图6中所示,催化剂b和c比催化剂a更具有活性。例如,在70℃、具有相同的铂负载量,催化剂c的乙炔转化率约为13%,尽管催化剂b的乙炔转化率小于3%。但是在相同的反应条件下,催化剂a的乙炔转化率可以忽略不计。另一方面,在测量的温度范围内,催化剂a、b和c的乙烯选择性相似,约为65%。在这些测量中,使用相同量的催化剂,500mg,总气体流量为76ml/min,乙炔2ml/min、氢气6ml/min、氮气2ml/min和氦气66ml/min。
图7示出了选择性乙炔加氢反应中催化剂a(还原后的pt/al2o3)(曲线800)、催化剂b(还原后的pt/zsm-5)(曲线820)和催化剂c(活化后的pt/zsm-5)(曲线840)的绿油选择性。显然,催化剂b和催化剂c的绿油产率远低于催化剂a,更重要的是,催化剂c的绿油产率为零或几乎为零。由于绿油产物的存在会严重抑制加氢催化剂活性,且降低催化剂的使用寿命,因此将绿油产率降低到接近零或零的水平是催化剂c的一大优点。
根据图6和图7中所示的催化结果,可以断定的是:对于乙炔选择性加氢成乙烯,催化剂b(还原后的pt/zsm-5)比催化剂a(还原后的pt/al2o3)更具有活性,具有相似的乙烯选择性和降低的绿油产率。然而,催化剂c(ptcx/zsm-5)对乙炔加氢显示出更高的催化活性,具有相似的乙烯活性和可忽略不计的绿油产率。总的来说,与催化剂a(例如标准pt/al2o3催化剂)相比,具有本发明特征的催化剂c在选择性乙炔加氢反应中,在活性和选择性方面表现出显著提高的催化性能,例如封装的铂碳化物纳米团簇对乙炔选择性加氢成乙烯具有活性和选择性。
图8示出了在250-620℃温度范围内,乙烷在催化剂b(还原后的pt/zsm-5)上的程序升温表面反应数据(曲线900)。乙烷(曲线910)、甲烷(曲线920)和btx(曲线930)的产物浓度(基于碳)由质谱仪(ms)监测,并绘制在y轴上。
图9显示了在250-620℃温度范围内,乙烷在催化剂c(活化后的pt/zsm-5)上的程序升温表面反应数据(曲线1000)。乙烷(曲线1010)、甲烷(曲线1020)和btx(曲线1030)的产物浓度(基于碳)由质谱仪(ms)监测,并绘制在y轴上。
图10显示了催化剂b的甲烷和btx产率作为反应温度的函数(分别为曲线1120和1125),以及催化剂c的甲烷和btx产率作为反应温度的函数(分别为曲线1140和1145)。该图是基于从图8和图9获得的数据而绘制的。
图8、图9和图10示出了催化剂c(ptcx/zsm-5)对乙烷芳构化具有高活性。对于催化剂b(pt/zsm-5),数据表明,起燃温度约为400℃,铂碳化物类在该温度处开始形成。如表征数据所表明,在乙烷芳构化过程中,pt类以pt碳化物纳米团簇的形式存在,因此需要初始引入期来活化催化剂b。而对于催化剂c,不需要引入期。因此,对于催化剂c,起燃温度约为300℃,该温度比催化剂b低得多。此外,催化剂c的初始btx选择性远高于催化剂b的btx选择性。这些观察结果结合表征数据清楚地表明,原位形成的限制的pt碳化物纳米团簇是pt/zsm-5基催化剂在乙烷芳构化反应中的活性相,例如,封装的pt碳化物纳米团簇对乙烷芳构化反应具有活性和选择性。
测量还表明,对于乙炔加氢和乙烷芳构化反应,在催化剂b和催化剂c的情况,惰性粘合剂与pt/zsm-5的混合不会改变催化剂活性和选择性。因此,惰性粘合剂不参与碳氢化合物转化的催化反应。
综上所述,数据表明,催化剂c(乙烷活化后的pt/zsm-5)形成了新型的封装的pt碳化物纳米团簇,其与先前的加氢催化剂相比具有许多优点。例如,在不限制本发明的情况下,其优点包括:
1.金属碳化物材料可以在环境压力下合成。
2.金属碳化物材料可以在300-650℃的温度下合成。
3.pt碳化物材料可以在不使用昂贵的金刚石载体和激光加热的情况下,通过使用市售的沸石载体和pt前体、优选地硝酸铂来合成。
4.金属碳化物可以在载体中形成为纳米团簇。
因此,本发明提供的上述实施方案至少包括以下三种方法:
1.用金属溶液浸渍沸石,随后煅烧、还原和乙烷活化来制备催化剂。
2.制备的催化剂在催化碳氢化合物转化反应中的高催化性能的说明。
3.制备的催化剂的活性相通过各种原位表征来鉴定。
上面的描述仅仅是本发明的各种实施方案,其中本发明的范围不限于此,并且本领域技术人员可以容易地做出本发明的技术范围内的改变和替换,并且应当认为这些改变或替换落入本发明的范围内。因此,本发明的保护范围仅以所附权利要求书的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!