Bi4V2O11/g-C3N4异质结光催化剂及其制备方法与应用与流程
本发明属于g-c3n4光催化剂技术领域。具体涉及一种bi4v2o11/g-c3n4异质结光催化剂及其制备方法与应用。
背景技术:
g-c3n4是一种半导体材料,能够作为光催化剂应用于有机污染物的光降解。g-c3n4的合成方法主要有热缩聚合成法,溶剂热合成法,电化学沉积法和固相合成法。其中热缩聚合成法主要是以尿素、三聚氰胺等为原料在400~600℃直接热缩聚得到。但以尿素为原料采用热缩聚合成法直接热缩聚得到的g-c3n4存在比表面积小,传质作用差,光生电子对复合严重等问题,影光催化活性。
bi系化合物如biobr,bimoo,biti2o,bipo4和bivo4等均可作为良好的光催化剂,bi系化合物催化剂与g-c3n4复合后能够有效的抑制光生电荷的复合,且g-c3n4的稳定性提高。因此,现有技术中常采用bi系化合物与g-c3n4复合,形成复合光催化剂。然而现有的复合光催化剂的制备方法中,通常先分别制备bi系化合物与g-c3n4,然后再将二者复合。其中bi系化合物的制备常采用传统的水热法处理。例如cn108325554a公开了一种钒酸铋/石墨相氮化碳符合材料、其制备方法及用途,其制备方法:将表面活性剂与特定铋离子浓度的水溶液混合,得到溶液a;将酸处理后的石墨相氮化碳与特定钒离子的水溶液混合,得到溶液b;将溶液a加入到溶液b中,并控制铋钒摩尔比,得到混合液;再进行水热反应,得到钒酸铋/石墨相氮化碳复合材料。因此,现有的复合光催化剂的制备方法较为复杂,制备成本高,不利于工业化推广应用。且催化活性仍然欠佳,因此有待改进。
另外,现有的复合光催化剂主要用于有机污染物的光催化降解,例如降解水中的有机污染物,而用于降解大气污染物中co2的复合光催化剂较少,且催化活性不高,导致co的产量偏低且采用。然而,近年来,化石燃料的不断开发和森林植被的严重破坏导致co2排放过量,从而使温室效应愈加严重,人类的生命安全受到更加大的威胁。为此,研发新型的g-c3n4结光催化剂以解决现有技术中的上述技术问题,十分必要。
技术实现要素:
本发明的第一个目的是提供一种bi4v2o11/g-c3n4异质结光催化剂,能够解决在光催化过程中的光响应范围窄、电子空穴复合率高和光催化活性低的技术问题。
为了实现上述目的,本发明采用如下技术方案:
bi4v2o11/g-c3n4异质结光催化剂,包括bi4v2o11和g-c3n4,其中,bi4v2o11占催化剂总重量的5~40%,其余为g-c3n4。
本发明制备的bi4v2o11/g-c3n4异质结光催化剂,通过bi4v2o11与g-c3n4的有效复合,得到的异质结光催化剂具有良好的光催化活性。
优选地,所述bi4v2o11占催化剂总重量的10~30%。
本发明的第二个目的是提供上述bi4v2o11/g-c3n4异质结光催化剂的制备方法,包括如下步骤:
(1)配方量的g-c3n4粉末,加入适量的乙二醇中并搅拌均匀;
(2)随后向步骤(1)的溶液中加入配方量的硝酸铋和钒酸铵,搅拌,得到均匀的溶液;
(3)将步骤(2)的溶液转入高压釜中,控制在150~200℃热处理5~15小时,然后将所得产物洗涤、烘干,得到bi4v2o11/g-c3n4异质结光催化剂;
所述bi4v2o11占催化剂总重量的5%~40%;所用的原料硝酸铋和钒酸铵中,铋元素和钒元素的摩尔比为1.5~2.5:1。
需要说明的是,本发明中,不限定所述g-c3n4粉末的制备方法和原料。可采用现有技术中常规的原料和方法制备所述g-c3n4粉末。
需要说明的是,所述洗涤步骤中,洗涤掉水溶性的杂质离子,并洗涤至ph中性。
优选地,采用去离子水和乙醇洗涤。更优选地,采用去离子水洗涤3次,然后采用乙醇洗涤3次。
根据本发明的一个优选技术方案,所述步骤(1)中,所述g-c3n4粉末的制备包括如下步骤:将尿素或三聚氰胺研磨0.1~10个小时,然后转移至坩埚内,并以5~20℃/分钟的速率加热至400~450℃,然后保温1~3小时;再以5~20℃/分钟的速率升温至500~600℃,然后再保温1~3小时。
进一步优选地,所述尿素或三聚氰胺研磨0.1~4个小时,然后转移至坩埚内,并以5~20℃/分钟的速率加热至400℃,然后保温1~3小时,再以5~20℃/分钟的速率升温至550℃,然后再保温1~3小时。上述条件下,所得的g-c3n4粉末具有更大的比表面积。
更优选地,所述尿素或三聚氰胺研磨0.1~2个小时,然后转移至坩埚内,并用铝箔纸包裹密封,然后以16℃/分钟的速率加热至400℃,然后保温2小时;再以16℃/分钟的速率升温至550℃,然后保温2小时。
上述条件下得到的g-c3n4粉末具有更大的比表面积,且研磨时间较短,经济效益高。
进一步优选地,控制研磨速率为30~40r/min。
优选地,所述步骤(1)中,所述g-c3n4粉末加入适量的乙二醇中,经过超声处理,使其分散均匀。在上述超声处理条件下,所述g-c3n4具有更好的溶解均匀性,有利于后续的催化剂制备,确保良好的催化活性。同时,超声处理后,g-c3n4会剥离成薄片状,也会增大其比表面积的大小。
优选地,所述超声处理的条件为:超声功率1000~3000w,超声10~60min。
优选地,所述步骤(1)中,所述乙二醇的体积与g-c3n4粉末的质量比为(100~200)ml:1g。
在上述条件下,原料可在乙二醇体系中得到良好的分散,确保bi4v2o11/g-c3n4异质结的形成,从而保证bi4v2o11/g-c3n4异质结光催化剂良好的催化性能。
优选地,所述步骤(2)中,所用的原料硝酸铋和钒酸铵中,铋元素和钒元素的摩尔比为1.5~2.0:1。在上述条件下,bi4v2o11/g-c3n4异质结光催化剂具有更好的催化性能。
优选地,所述步骤(2)中,控制搅拌速率为400~600r/min,以获得均匀的溶液;所述步骤(3)中,控制在170~190℃处理7~15小时。
优选地,所述步骤(3)中,所述烘干温度为60~100℃,烘干时间为12~24h。
更优选地,所述烘干温度为70℃,烘干时间为20h。
本发明的第三个目的是提供上述bi4v2o11/g-c3n4异质结光催化剂的应用,用于co2的催化还原。
优选地,所述co2催化还原的催化产物主要为co。
与现有技术相比,本发明具有如下有益技术效果:
(1)、本发明的bi4v2o11/g-c3n4异质结光催化剂,铋系化合物bi4v2o11与g-c3n4构建异质结,有效降低了电子空穴对复合率;且比表面积高,具有优异的可见光催化活性,同时稳定性好,且催化寿命长。
(2)、本发明的制备方法,通过特定配比的g-c3n4粉体、硝酸铋和钒酸铵在乙二醇体系下,在特定的温度和时间条件下,在高压釜中发生溶剂热反应,得到掺杂了bi4v2o11的bi4v2o11/g-c3n4异质结光催化剂,不仅工艺简单,易于操作,且所用原料硝酸铋、钒酸铵与g-c3n4粉体均容易获得,成本低,适合大规模工业化生产。
(3)、本发明的bi4v2o11/g-c3n4异质结光催化剂用于co2催化还原时,具有优异的还原性能,对于co产量提升效果明显。
附图说明
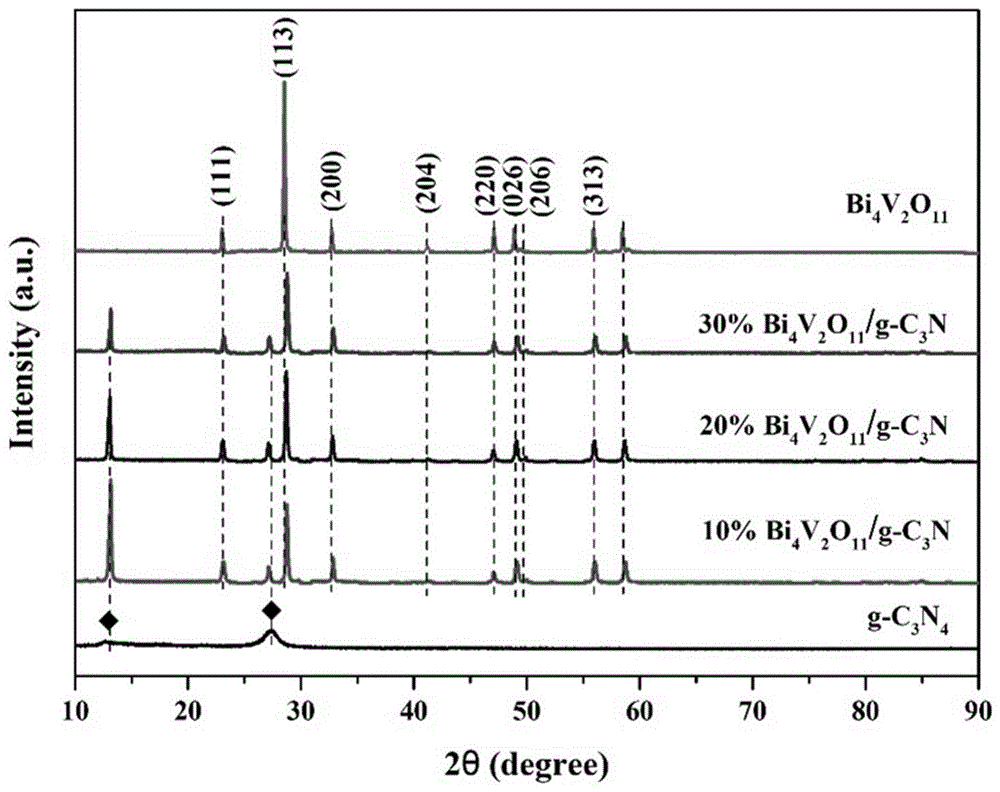
图1为实施例2-4的bi4v2o11/g-c3n4异质结光催化剂,及bi4v2o11标准样品、g-c3n4粉末的xrd图谱。
图2为co2光催化还原反应8小时的结果。其中横坐标为时间,纵坐标表示co转化产量。
具体实施方式
以下结合具体实施例,对本发明的bi4v2o11/g-c3n4异质结光催化剂及其制备方法作进一步说明。应理解,以下实施例仅用于说明本发明而非用于限定本发明的范围。对于未注明的工艺参数,可参照常规工艺进行。
实施例1、制备g-c3n4粉体
g-c3n4粉体1:称量25g尿素放入玻璃研钵中,研磨2个小时,控制研磨速率为30~40r/min;然后转移至氧化铝坩埚中用铝箔纸包裹密封,以16℃/min的速率加热至400℃,然后保温2小时;接着用相同的加热速率升高至550℃,然后再保温2小时;最后将坩埚自然冷却至室温,所得浅黄色粉末即g-c3n4粉体。所述的g-c3n4粉体呈卷曲的片状。
g-c3n4粉体2:主要的制备步骤与g-c3n4粉体1相同,区别在于:研磨4个小时。
g-c3n4粉体3:主要的制备步骤与g-c3n4粉体1相同,区别在于:不经过研磨。
g-c3n4粉体4:采用三聚氰胺为原料,以10℃/min的速率加热至520℃,保温4小时。
实施例2、10%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂
bi4v2o11/g-c3n4异质结光催化剂的制备包括如下步骤:
(1)、称量0.5gg-c3n4粉体1,将其加入60ml乙二醇中,并超声处理30分钟,超声功率为1800w,获得含有纳米片状g-c3n4的溶液。
(2)、随后向步骤(1)的上述溶液中加入0.095g的bi(no3)3·5h2o和0.012g的nh4vo3,剧烈搅拌30分钟以获得均匀溶液,搅拌速率为500r/min。
(3)、将步骤(2)的均匀溶液转入聚四氟乙烯衬里的高压釜中,控制在180℃热处理8小时,然后将所得产物用去离子水洗涤3次,然后用乙醇洗涤3次,并在70℃下烘干20h,最终获得10%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂a。
对bi4v2o11/g-c3n4异质结光催化剂a进行xrd检测,结果如图1所示。由图1可以看出,实施例2制备的bi4v2o11/g-c3n4异质结光催化剂a包括bi4v2o11和g-c3n4。
实施例3、20%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂
实施例3的基本步骤与实施例2相同,区别在于:
步骤(2)中,加入0.19g的bi(no3)3·5h2o和0.024g的nh4vo3。
步骤(3)中,在180℃热处理12小时。
最终获得20%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂b。
对bi4v2o11/g-c3n4异质结光催化剂b进行xrd检测,结果如图1所示。由图1可以看出,实施例3制备的bi4v2o11/g-c3n4异质结光催化剂b包括bi4v2o11和g-c3n4。
实施例4、30%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂
实施例4的基本步骤与实施例2相同,区别在于:
步骤(2)中,加入0.285g的bi(no3)3·5h2o和0.036g的nh4vo3。
步骤(3)中,在180℃热处理12小时。
最终获得30%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂c。
对bi4v2o11/g-c3n4异质结光催化剂c进行xrd检测,结果如图1所示。由图1可以看出,实施例4制备的bi4v2o11/g-c3n4异质结光催化剂c包括bi4v2o11和g-c3n4。
实施例5
实施例5的基本步骤与实施例3相同,区别在于:
步骤(1)中,采用g-c3n4粉体2。
最终获得20%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂d。
bi4v2o11/g-c3n4异质结光催化剂f的xrd检测结果与实施例3类似。
实施例6
实施例6的基本步骤与实施例3相同,区别在于:
步骤(1)中,采用g-c3n4粉体3。
最终获得20%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂e。
bi4v2o11/g-c3n4异质结光催化剂g的xrd检测结果与实施例3类似。
实施例7
实施例7的基本步骤与实施例3相同,区别在于:
步骤(1)中,采用g-c3n4粉体4。
最终获得20%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂f。
bi4v2o11/g-c3n4异质结光催化剂g的xrd检测结果与实施例3类似。
实施例8
实施例8的基本步骤与实施例3相同,区别在于:
所述步骤(2)中,加入0.19g的bi(no3)3·5h2o和0.031g的nh4vo3。
最终获得bi4v2o11/g-c3n4异质结光催化剂g。
bi4v2o11/g-c3n4异质结光催化剂g的xrd检测结果与实施例3类似。
实施例9
实施例9的基本步骤与实施例3相同,区别在于:
所述步骤(2)中,加入0.19g的bi(no3)3·5h2o和0.018g的nh4vo3。
最终获得bi4v2o11/g-c3n4异质结光催化剂h。
bi4v2o11/g-c3n4异质结光催化剂g的xrd检测结果与实施例3类似。
实施例10
实施例10的基本步骤与实施例3相同,区别在于:
步骤(1)中,乙二醇的用量为100ml,无超声处理。
步骤(3)中,在150℃热处理15小时。
最终获得20%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂i。
bi4v2o11/g-c3n4异质结光催化剂d的xrd检测结果与实施例3类似。
实施例11
实施例11的基本步骤与实施例3相同,区别在于:
步骤(1)中,乙二醇的用量为50ml。
步骤(3)中,在200℃热处理5小时。
最终获得20%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂j。
bi4v2o11/g-c3n4异质结光催化剂e的xrd检测结果与实施例3类似。
实施例12
实施例12的基本步骤与实施例3相同,区别在于:
步骤(3)中,在190℃水热处理7小时。
最终获得20%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂k。
bi4v2o11/g-c3n4异质结光催化剂i的xrd检测结果与实施例3类似。
实施例13
实施例13的基本步骤与实施例3相同,区别在于:
步骤(3)中,在170℃水热处理9小时。
最终获得20%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂l。
bi4v2o11/g-c3n4异质结光催化剂j的xrd检测结果与实施例3类似。
实施例14
实施例14的基本步骤与实施例2相同,区别在于:
bi4v2o11/g-c3n4异质结光催化剂的制备包括如下步骤:
(1)、称量2gg-c3n4粉体1,将其加入240ml乙二醇中,并超声处理30分钟,超声功率为1800w,获得含有纳米片状g-c3n4的溶液。
(2)、随后向步骤(1)的上述溶液中加入0.038g的bi(no3)3·5h2o和0.005g的nh4vo3,剧烈搅拌30分钟以获得均匀溶液,搅拌速率为500r/min。
(3)、将步骤(2)的均匀溶液转入聚四氟乙烯衬里的高压釜中,控制在180℃热处理8小时,然后将所得产物用去离子水洗涤3次,然后用乙醇洗涤3次,并在70℃下烘干20h,最终获得1%bi4v2o11掺杂的bi4v2o11/g-c3n4异质结光催化剂m。
bi4v2o11/g-c3n4异质结光催化剂k的xrd检测结果与实施例2类似。
实施例13、co2光催化还原反应
co2光催化还原反应在500ml反应器气体封闭循环系统labsolar-6a中进行,labsolar-6a购买自北京泊菲莱科技有限公司(perfectlightcompany)。
在反应过程中,用300w氙弧灯为光源,在充分搅拌下将50mg实施例2-12、及g-c3n4粉体1的光催化剂均匀分散至100ml去离子水中,形成悬浮液。照明前,将悬浮液进行真空处理(0-1kpa),co2(99.999%)用质量流量计控制流量,均匀通入所述悬浮液中,压力为100kpa,并保持30分钟,以达到吸附-解吸平衡,然后放上光源进行实验。
反应器温度和压力分别保持在25℃和100kpa。在反应过程中,每小时从玻璃室采样0.15ml气体,并通过气相色谱(gc-2010plus,shimadzu,日本)检测碳基气体产物co的含量。
实施例2-4及g-c3n4粉体1的对比结果如图2所示。由图2可以看出,相同的反应条件及反应时间前提下,实施例2-4的光催化剂a-c的co产量均明显比g-c3n4粉体1高,且20%的bi4v2o11掺杂量的光催化剂co产量最高。
选择反应时间为6小时时实施例2-12的光催化剂的co2光催化还原反应数据及光催化剂的性质列于表1,以进一步进行比较。
表1co2光催化还原反应6小时的结果μmol/g
由表1的数据可知,与g-c3n4光催化剂相比,本发明制备的bi4v2o11/g-c3n4异质结光催化剂,其co产量明显提高,因而co2催化还原反应的活性高。可能的原因是:比表面积的增大和催化剂异质结内部电场的建立,可减小电子空穴对复合率,从而提高催化剂的活性。另外,催化剂稳定性好,活性增长稳定,没有明显的活性下降,因此使用寿命较长。
以上对本发明的具体实施例进行了详细描述,但其只作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对该实用进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!