一种加氢精制催化剂及其制备方法和应用与流程
1.本发明涉及一种加氢精制催化剂及其制备方法和应用,该催化剂特别适用于页岩油馏分油加氢处理工艺。
背景技术:
2.在自然界的资源中,油页岩和石油一样主要由藻类等低等浮游生物经腐化作用和煤化作用而生成。通过低干馏等办法,从油页岩中“榨”出的页岩油则被称作“人造石油”,经过进一步加工提炼后可以制得汽油、煤油、柴油等液体燃料。在油页岩的开采初期由于技术不过关,生产过程污染环境,这一行业发展受到限制。近年来,随着技术的进步,该问题已得到了很好的解决。可以预见,在当前石油资源紧缺、油价高涨的形势下,页岩油将在能源家族中扮演越来越重要的角色。但与天然石油不同的是,页岩油中含有更多的不饱和烃以及硫、氮、氧等非烃组分,页岩油中高含量杂环芳烃大大限制了它直接作为运输燃料油,大量硫、氮杂质产生的nox、sox,对环境产生不利影响。
3.页岩油的加工手段主要有非加氢处理和加氢处理两种方法。非加氢处理一般包括酸碱精制、溶剂精制、吸附精制和加入稳定剂等。加氢处理方法方面,美国炼油公司主要是对页岩油进行加氢预处理以脱除其中硫、氮、砷等杂质,然后在炼厂按常规加工工艺生产各种油品;巴西炼油公司将页岩油分为轻、重两种馏分,轻馏分经催化裂化生产汽油产品,重馏分则作为燃料油;澳大利亚spp公司对页岩油进行加氢精制以生产超低硫轻质燃料油。
4.加氢处理过程就是用含有周期表中第
ⅷ
族及第ⅵb族的金属氧化物负载到耐熔无机多孔材料中,一般采用氧化铝、氧化硅、二氧化钛、碳化硅、氧化硼、氧化锆以及它们组合在一起的复合型载体,通过浸渍工艺过程,制备出催化剂前体,再经过若干步的干燥和焙烧工序制备出成品催化剂。成品催化剂在使用前进行预硫化,即在含有硫化氢、含硫有机化合物或者是单质硫存在的情况下,使氧化态催化剂转化成硫化态催化剂,进行加氢反应。
5.usp4419218公开了一种用脱金属后页岩油加氢裂化生产航煤的方法,其中加氢精制剂是以mo-ni-p为活性金属组分,以氧化铝为载体,加氢裂化催化剂是以co-cr-mo三金属为活性组分,以zsm-12 分子筛为载体,航煤产率达到70%,但精制剂效果较差,进而影响裂化产品的质量。
6.cn1785512a公开了一种含亚铁的烃类裂化催化剂制备方法,该催化剂是由5~20%的磷酸氢二铝、5~15%的亚铁,余量为粘土组成,适用于固定床高氮页岩油和高含蜡原油在固定床中的烃类催化裂化反应,裂化性能良好。
7.cn101590416a公开了一种钼镍加氢催化剂的制备方法,该方法是采用混捏-浸渍两个步骤进行制备催化剂,首先将氧化钼、含钛化合物、含磷化合物加入到氧化铝和/或氧化铝前身物中,再加入硝酸溶液,混捏,挤条成型,干燥、焙烧得到含钛、磷、钼的氧化铝成型物,再浸渍含镍的磷酸溶液,经过干燥、焙烧后得到钼镍加氢催化剂,但该催化剂对于含氮量高的馏分油精制效果不佳。
8.cn1052501a公开了一种加氢精制催化剂及制法,该催化剂以氧化硅-氧化铝为载
体,采用w-mo-ni三种活性金属组分和硼助剂,采用分段浸渍法浸渍,经过干燥、焙烧后得到成品催化剂,氮含量增加时,脱氮效果不明显。
9.现有技术制备的加氢处理催化剂脱氮活性低,尤其是遇到含氮量高的页岩油馏分油,脱氮效果不佳,直接会影响后续裂化段反应器中催化剂的反应性能,进而影响裂化产品的质量,缩短装置运转周期,增加了运行成本。
技术实现要素:
10.为了克服现有技术中的不足之处,本发明提供了一种适用于处理页岩油的加氢精制催化剂及其制备方法和应用。本发明的加氢精制催化剂能够深度脱除页岩油中的含氮化合物,加氢性能好,满足后续工序生产的质量要求,确保充分发挥裂化催化剂的使用性能。
11.本发明第一方面提供了一种加氢精制催化剂,所述加氢精制催化剂包括:载体、活性金属组分、纳米氧化镁和有机助剂,其中载体包括al-sba-15分子筛和氧化铝。
12.进一步地,所述加氢精制催化剂的性质如下:比表面积为180~240m2/g,孔容为0.28~0.45ml/g,中强酸含量占总酸含量的50%~70%,优选53%~63%。
13.所述活性金属组分为第
ⅷ
族金属和第ⅵb族金属,第
ⅷ
族金属优选为co和/或ni,第ⅵb族金属优选为w和/或mo。以最终加氢精制催化剂的重量为基准,第
ⅷ
族金属以氧化物计的含量为1wt%~15wt%,优选3wt%~10wt%,第ⅵb族金属以氧化物计的含量为10wt%~30wt%,优选15wt%~25wt%。
14.进一步地,所述加氢精制催化剂中,以催化剂的重量计,载体的含量为60%~74%,活性金属组分以氧化物计的含量为11%~31%,纳米氧化镁的含量为1%~6%。
15.进一步地,所述加氢精制催化剂中,有机助剂的含量与第ⅵb族金属原子摩尔比为0.01:1~12:1,优选为0.01:1~10:1。
16.进一步地,所述有机助剂为核糖醇、d-甘露糖醇、水苏糖中的一种或几种的组合。
17.进一步地,所述的载体中,al-sba-15分子筛的重量含量为2%~20%,优选为3%~12%,氧化铝的重量含量为80%~98%,优选为88%~97%。
18.进一步地,所述al-sba-15分子筛的孔分布包括:孔直径《4nm的孔所占的孔容为总孔容的20%以下,优选15%以下;所述al-sba-15分子筛中,b酸与l酸的比值在1以下。
19.进一步地,所述al-sba-15分子筛中b酸与l酸的比值可以为在0.8以下,也可以为在0.5以下,还可以为在0.4以下。所述分子筛中b酸与l酸的比值可以在0.1以上,也可以为0.2以上。
20.进一步地,所述al-sba-15分子筛中,中强酸酸量为0.6~1.0ml/g,优选为0.7~0.9ml/g。
21.进一步地,所述al-sba-15分子筛中,氧化铝的质量含量为2%~85%,优选为5%~82%,进一步优选为5%~75%。所述分子筛中,氧化铝的含量可以在宽范围内调整,比如可以为10%,15%,16%,18%,20%,25%,30%,32%,35%,40%,45%,50%,55%,60%,70%,75%等。
22.进一步地,所述al-sba-15分子筛的孔分布还包括:孔直径为4~15nm的孔所占的孔容为总孔容的40%~70%,优选为45%~65%,进一步优选为50%~60%。
23.进一步地,所述al-sba-15分子筛的性质如下:比表面积为550~850m2/g,优选为650~750m2/g,总孔容为0.7~1.3ml/g,优选为0.9~1.2ml/g。
24.本发明第二方面提供了一种加氢精制催化剂的制备方法,包括:(i)以无定形硅铝干胶为原料,采用p123三嵌段共聚物为模板剂制备al-sba-15分子筛;(ii)将步骤(i)制备的 al-sba-15介孔分子筛、氧化铝、水混合制成浆液;(iii)将含活性金属组分的溶液和含纳米氧化镁分散液加入到步骤(ii)制备的浆液中,得到混合浆液;(iv)将步骤(iii)制备的混合浆液经过滤、洗涤、热处理后,得到第一催化剂前驱体;(v)将步骤(iv)制备的第一催化剂前驱体进行粉碎,混捏,成型,干燥,焙烧得到第二催化剂前驱体;(vi)将步骤(v)制备的第二催化剂前驱体用含有机助剂的水溶液进行浸渍,浸渍后的样品经热处理后,得到最终加氢精制催化剂。
25.进一步地,步骤(i)制备al-sba-15分子筛的方法,包括:(1)将无定形硅铝干胶和水混合形成浆液;(2)配制含有p123三嵌段共聚物的酸性溶液;(3)将步骤(1)制备的浆液与步骤(2)配制的含有p123三嵌段共聚物的酸性溶液混合;经过晶化,制得al-sba-15分子筛。
26.进一步地,所述无定形硅铝干胶中,氧化铝的质量含量为2%~85%,优选5%~82%,进一步优选5%~75%。氧化铝的质量含量可以在宽范围内调整,比如可以为10%,15%,16%,18%,20%,25%,30%,32%,35%,40%,45%,50%,55%,60%,70%,75%等。
27.进一步地,所述无定形硅铝干胶的性质如下:比表面积为400~650m2/g,优选450~600m2/g,孔容为0.52~1.8ml/g,优选0.85~1.5ml/g,孔分布如下:孔直径为4~15nm的孔容占总孔容的85%~95%,孔直径>15nm的孔容占总孔容的5%以下。
28.进一步地,步骤(1)中所述无定形硅铝干胶采用碳化法制备,可以采用如下过程制备: a、分别配制铝酸钠溶液和硅酸钠溶液;b、向铝酸钠溶液中加入部分或全部硅酸钠溶液,然后通入co2气体,控制反应温度为10~40℃,优选为15~35℃,控制成胶的ph值为8~11;其中当通入的co2气体量占总通入量的40%~100%,优选为50%~80%时,加入剩余硅酸钠溶液;c、在步骤b的控制温度和ph值下,上述混合物通风稳定10~30分钟;d、将步骤c所得的固液混合物过滤,滤饼洗涤;e、将步骤d所得的滤饼打浆,然后进行水热处理,经过滤、干燥,得到所述的无定形硅铝干胶;所述的水热处理条件如下:在120~150℃,0.5~4.0mpa水蒸汽压力下处理2~10小时。
29.进一步地,步骤a中,铝酸钠溶液的浓度为15~55gal2o3/l,进一步可以为15~35gal2o3/l,硅酸钠溶液的浓度为50~200gsio2/l,进一步可以为50~150gsio2/l。
30.进一步地,步骤b中加入部分或全部的硅酸钠溶液,即为所加入的全部硅酸钠溶液的5wt%~100wt%。所述co2气体的浓度为30v%~60v%。在步骤b成胶过程中通风搅拌。
31.进一步地,步骤b的具体过程为下面几种情况:(1)向铝酸钠中加入全部硅酸钠后,通入co2气体;(2)向铝酸钠中加入部分硅酸钠后,通入全部co2气体,然后向混合物中加入剩
余硅酸钠溶液;(3)向铝酸钠中加入部分硅酸钠后,通入部分co2气体,再一边通co2气体一边加入剩余硅酸钠溶液。
32.进一步地,将步骤d所得的浆液过滤并用50~95℃去离子水洗至接近中性,进一步地,将步骤e所得滤饼按固液体积比为8:1~12:1,加水打浆。
33.进一步地,步骤e所述的干燥可采用常规方法进行,可以在110~130℃下干燥6~8小时。
34.进一步地,步骤(1)中所述的无定形硅铝干胶与水的质量比为10:90~30:70,优选15:85~25:75。
35.进一步地,步骤(2)中所述酸性溶液的ph为1~5,优选1.2~2.3,所述酸性水溶液中p123三嵌段共聚物的质量含量为 0.5%~5.0%,优选0.8%~2.8%。
36.进一步地,步骤(2)中将p123三嵌段共聚物加入到稀酸(比如稀盐酸)中,所述的稀酸溶液的浓度以h
+
计为0.05~0.3mol/l,优选0.1~0.2 mol/l,进一步优选0.13~0.18 mol/l;为了使p123三嵌段共聚物充分溶解,温度体系控制为10~60℃,优选20~40℃,进一步优选25~35℃。
37.进一步地,步骤(3)中将步骤(1)制备的浆液同步骤(2)配制的含有p123三嵌段共聚物的酸性水溶液混合,步骤(1)制备的浆液和步骤(2)配制的含有p123三嵌段共聚物的酸性水溶液的量以混合后的体系中p123三嵌段共聚物和无定形硅铝的质量比为0.5:1~5:1,优选1:1~5:1,进一步优选1:1~3:1。
38.进一步地,步骤(3)中所述的晶化温度为80~120℃,优选90~110℃;晶化时间为10~35h,优选16~24h;晶化过程中ph控制为2.0~5.0,优选3.2~4.8。
39.进一步地,在步骤(3)晶化步骤结束后,可以通过常规已知的任何方式从所获得的混合物中分离出al-sba-15分子筛,比如采用过滤、洗涤和干燥等至少一个步骤。所述的过滤,可以采用抽滤。所述的洗涤,可以采用去离子水为洗涤液来进行洗涤。所述的干燥,可以在80~150℃,优选90~130℃,干燥时间为2~12h,优选 3~6h。该干燥可以在常压下进行。
40.进一步地,根据需要,还可以将上述方法制备的分子筛进行焙烧,以脱除模板剂和可能存在的水分等。所述焙烧可以按照本领域常规已知的任何方式进行,比如焙烧温度一般为450~600℃,优选480~580℃,进一步优选500~560℃,所述焙烧时间为2~10h,优选 3~6h。另外,所述焙烧一般在含氧气氛下进行,比如空气或者氧气气氛下。
41.进一步地,步骤(ii)中所述氧化铝的性质如下:比表面积为150~450m2/g,优选为230~340m2/g;孔容为0.4~1.4ml/g,优选为0.8~1.2ml/g,平均孔直径为8~14nm。
42.进一步地,步骤(ii)中,所述的含纳米氧化镁的分散液中纳米二氧化镁的质量浓度为20%~50%。所述含纳米二氧化镁的分散液可采用市售的纳米二氧化镁分散液,所述纳米氧化镁的粒径为20nm~40nm。
43.进一步地,将步骤(iii)制备的两种溶液同时或者分别加入到步骤(ii)制备的浆液中;进一步地,将步骤(iii)制备的混合浆液在超声波作用下进行搅拌处理,超声波的功率为5~100hz,优选为20~60 hz;温度控制在20~80℃,优选为40~60℃,搅拌时间为10~80min,优选为20~60min。
44.进一步地,步骤(iv)中,所述过滤、洗涤采用本领域的常规手段,所述热处理的温
度为60~180℃,时间为0 .5~20h,优选为60~120℃,时间为1~8h,所述热处理在氮气或惰性气体保护下进行。
45.进一步地,步骤(v)中,所述的混捏、挤条采用本领域的常规手段,混捏或成型过程中可加入常规的助剂,比如胶溶酸、助挤剂、粘合剂等,胶溶酸可以为柠檬酸和硝酸中的至少一种,优选柠檬酸和硝酸。粘合剂可以为小孔氧化铝。助挤剂可以为田菁粉等。
46.进一步地,步骤(v)中,所述的干燥条件为:干燥温度为60℃~220℃,优选为90℃~180℃,干燥时间为0.5h~10h,优选为1h~5h。所述的焙烧条件为:所述焙烧条件如下:焙烧温度为350℃~500℃,优选为400℃~480℃,焙烧时间为0.5h~10h,优选为1h~5h。
47.进一步地,步骤(vi)所述的有机助剂为核糖醇、d-甘露糖醇、水苏糖中的一种或几种的组合。
48.进一步地,步骤(vi)中,含有机助剂的水溶液为将有机助剂充分溶解于水中即可。
49.进一步地,步骤(vi)所述热处理优选为两步热处理,其中,第一步热处理的温度为80~120,优选为90~110℃,时间为0.5~5h,优选为1~3h,第二步热处理温度为120~200℃,优选为150~180℃,时间为0.5~5h,优选为1~3h。
50.进一步地,所述加氢精制催化剂中,还可以含有常规助剂,如p、b、ti、zr等中的至少一种,其中,助剂的含量以催化剂重量计为所述加氢精制催化剂重量的10%以下,可以为0.1%~8.0%。
51.本发明还提供了一种所述的加氢精制催化剂的应用。
52.进一步地,所述应用为将所述加氢精制催化剂应用于页岩油加氢脱氮反应。
53.进一步地,所述加氢精制催化剂应用于页岩油加氢脱氮反应的反应条件如下:反应总压8~16mpa,液时体积空速0.2~8.2h-1
,氢油体积比500:1~1500:1,反应温度为350~400℃。所述的页岩油的性质如下:氮含量>1wt%,硫含量>0.5wt%,氧含量>0.8wt%,且与常规原油相比,本技术所述的页岩油所含的不饱和芳烃以及杂质、金属含量高。
54.同现有技术相比,本发明所述的加氢精制催化剂及其制备方法具有如下优点:(1)本发明加氢精制催化剂载体所采用的al-sba-15分子筛,可根据原料的特点需求调整al-sba-15分子筛的酸量。该分子筛的加入能够显著改善催化剂酸性质,强酸含量减少,中强酸含量显著增加,催化剂的本征活性得到很好改善;其次,本发明的al-sba-15分子筛即使是在铝含量非常高(比如氧化铝在分子筛化学组成中所占的质量百分比含量高于7重量%)的情况下,分子筛仍然显示出介孔结构的规整性,该规整性可以用分子筛的孔分布(特别是孔直径《4nm的孔的孔容比例)进行表征。作为佐证,根据本发明的al-sba-15分子筛,即使氧化铝在分子筛化学组成中所占的质量百分比含量在2%至85%之间广泛变化,孔直径《4nm的孔所占的孔容仍然为总孔容的20%以下,保持了介孔结构的完整性和规整性,这是现有技术所制造的al-sba-15分子筛所不具备的。由此加入该al-sba-15分子筛之后催化剂载体的孔道结构会向介孔方向迁移,有益于页岩油中大分子稠环芳烃反应;载体中al-sba-15介孔分子筛和氧化铝在使用性能上相互协调,产生较好的协同催化作用,加入al-sba-15介孔分子筛可以显著提高活性金属组分在载体表面的浓度,即活性金属组分的分散度增加,有益于产生更多的活性位,提高催化剂的反应活性。
55.(2)本发明的加氢精制催化剂中还含有有机助剂,其能进一步优化催化剂的酸性
质,催化剂的脱氮活性明显增加;同时,加入有机助剂能够削弱活性组分与载体间的作用力,活性组分易于还原,有机助剂表面的大量羟基能够与活性组分络合,进一步增加了催化剂的有效活性位,提升催化剂综合使用性能。
56.(3)本发明的加氢精制催化剂中还含有纳米氧化镁,能够优化催化剂表面酸性,强酸量减少,中强酸和弱酸量适宜,提高催化剂的抗水能力,长时间运转后,催化剂的强度几乎不变。同时,催化剂脱氮活性得到很好地改善,延长装置的运转周期,降低运行成本。
附图说明
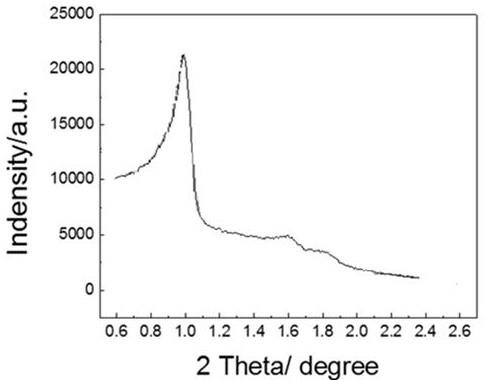
57.图1为本发明实施例1得到的al-sba-15分子筛的xrd图谱。
具体实施方式
58.本发明中,al-sba-15分子筛是指在sba-15分子筛中引入铝原子,而铝原子在sba-15分子筛中的存在状态并没有特别限制,一般部分铝原子分布在sba-15分子筛的骨架上。
59.本发明中,所述l酸或b酸的测定是采用红外光谱法,仪器是采用美国nicot傅里叶红外光谱仪-6700,测定方法如下:称取粒度小于200目的样品20mg压成直径为20mm的薄片,装在吸收池的样品架上,将200mg样品放入仪器吊杯中,连接好吸收池和吸附管,抽真空处理,真空度达到4
×
10-2
pa时,升温至500℃,保持1小时,以除去样品表面吸附物,降至室温后,吸附吡啶至饱和,继续升温至160℃,平衡1小时,脱附物理吸附的吡啶,进而可得到红外总酸、b酸和l酸的酸量,其中,b酸和l酸的酸量单位为mmol/l。
60.本发明中,中强酸酸量是采用nh
3-tpd方法测得。采用仪器为麦克仪器公司auto-chem ii 2920型化学吸附仪。采用氨气作为吸脱附介质,氦气作为载气,采用程序升温脱附和色谱分析得到不同脱附温度区的酸量,其中弱酸的酸量对应的氨气脱附温度为150℃~250℃中强酸的酸量对应的氨气脱附温度为250℃~400℃,强酸的酸量对应的氨气脱附温度为400℃~450℃,酸量单位:ml/g即每克分子筛吸附的氨气量。弱酸、中强酸和强酸的和为总酸量。
61.本发明中,所述比表面积、孔容和孔分布是采用asap2405物理吸附仪测定,测定方法如下:样品经过处理后,液态n2作吸附质,吸附温度为-196℃,进行分析测试。其中比表面积按bet法计算而得,孔容和孔分布是根据bjh法计算而得。
62.本发明中,所述xps表征金属分散度是采用美国thermo公司multilab2000x光电子能谱仪测得。mgkα作为激发源,能量为1253.6 ev,功率为200 w。以污染碳峰的c1s(284.6 ev)为定标标准,扣除荷电效应的影响,确定样品的真实结合能。
63.本发明中,xrd的测量是采用日本理学公司生产的d/max2500型x光衍射仪,测试条件如下:电压40kv,电流80ma,选用cukα靶,入射波长0.15405 nm。
64.下面通过实施例和对比例进一步说明本发明技术方案的作用及效果,但不应认为本发明仅局限于此具体的实施例中,本发明以下实施例及对比例中如无特殊的说明,百分数为质量分数。
65.实施例1(一)al-sba-15分子筛的制备(1)无定形硅铝干胶a1及浆液的制备:铝酸钠溶液浓度20gal2o3/l,硅酸钠溶液浓度
60gsio2/l,取0.75l铝酸钠溶液放入于成胶罐中,然后加入0.35l硅酸钠溶液,控制反应温度20℃,通入浓度为40v%的co2气体,通入co2气体占总通入量的50%时,一边通气一边加入0.20l硅酸钠溶液,控制成胶的ph值为9.7,然后通风稳定20分钟,浆液过滤并用65℃去离子水洗洗至中性,滤饼按固液体积比为12∶1加水打浆,在120℃,3.5mpa水蒸汽压力下处理2小时,在120℃干燥6小时后,粉碎过筛得无定形硅铝产品a1。将所制备的无定形硅铝a1和去离子水混合、打浆形成浆液;其中,无定形硅铝干胶与水的质量比为23:77;(2)配制含有p123三嵌段共聚物的酸性溶液;将p123三嵌段共聚物加入到稀盐酸中,所述的稀盐酸溶液的浓度为0.13mol/l,含有p123三嵌段共聚物的酸性水溶液的ph为1.3,含有p123三嵌段共聚物的酸性水溶液的温度为25℃,所述含有p123三嵌段共聚物的酸性水溶液中p123三嵌段共聚物的质量含量为1.6wt%;(3)将步骤(1)制备的浆液同步骤(2)配制的含有p123三嵌段共聚物的酸性水溶液混合,经过晶化、过滤、干燥和焙烧后制得al-sba-15分子筛,编号为a-s-1,混合后的体系中p123三嵌段共聚物和无定形硅铝的质量比为1.2:1,所述的晶化温度为90℃,晶化时间为20h;晶化过程中ph控制为3.3,所述干燥温度控制在100℃,干燥时间为3h,所述焙烧温度控制在550℃,所述焙烧时间为3h,a-s-1分子筛性质见表1。实施例1得到的a-s-1分子筛的xrd图谱见图1,显示出al-sba-15分子筛的特征峰。
66.(二)配置钼镍磷杂多酸溶液a,溶液的组成moo3的含量为45.5g/100ml,nio含量为7.5g/100ml,p的含量为3.5g/100ml,称取纳米氧化镁分散液10克(杭州万景新材料有限公司生产的产品,分散液中氧化镁含量30wt%,粒径30nm),用去离子水稀释至100ml,得到溶液b;称取a-s-1分子筛8g,氧化铝干胶粉92g放入6l去离子水中搅拌溶解,使两种粉体均匀分散在体系中,得到浆液c,将溶液a和b并流加入到浆液c中,在超声波功率52hz条件下搅拌处理,温度45℃,处理40min后,得到混合浆液,然后经过过滤、洗涤,然后再在氮气保护下110℃下热处理4小时,得到含纳米氧化镁以及活性组分的第一催化剂前驱体;将第一前驱体,粉碎后加入田菁粉4g,放入碾压机中,将柠檬酸4g,硝酸12.3g(浓度65%)溶解在125ml的去离子水里,制成酸性溶液,均匀倒入碾压机,经碾压25min后,物料呈膏状,在挤条机上挤成1.7mm三叶草,130℃干燥4小时,450℃焙烧3小时,得到第二催化剂前驱体;用含有水苏糖的水溶液浸渍第二催化剂前驱体,其中水苏糖的用量与mo原子摩尔比为1:1,浸渍后的样品室温晾干后,120℃热处理3h,150℃热处理1h,得到最终成品催化剂c-1,主要物理性质见表2和表3。
67.实施例2(一)al-sba-15分子筛的制备(1)无定形硅铝干胶a2的制备:铝酸钠溶液浓度30gal2o3/l,硅酸钠工作溶液浓度90gsio2/l,取1.25l铝酸钠溶液置于成胶罐中,然后加入0.65l硅酸钠溶液,控制反应温度32℃,通入浓度为52v%的co2气体,当ph值达到9.9时停止,然后通风稳定20分钟,洗至中性,滤饼按固液体积比为9∶1加水打浆,在130℃,3.9mpa水蒸汽压力下处理3小时,在130℃干燥8小时后,粉碎过筛得无定形硅铝产品a2。将所制备的无定形硅铝a2和去离子水混合、打浆形成浆液;其中,无定形硅铝干胶与水的质量比为25:75;(2)配制含有p123三嵌段共聚物的酸性水溶液;将p123三嵌段共聚物加入到稀盐酸中,所述的稀盐酸溶液的浓度为0.16mol/l,含有p123三嵌段共聚物的酸性水溶液的ph为1.8,
含有p123三嵌段共聚物的酸性水溶液的温度为33℃,所述含有p123三嵌段共聚物的酸性水溶液中p123三嵌段共聚物的含量为2.0wt%;(3)将步骤(1)制备的浆液同步骤(2)配制的含有p123三嵌段共聚物的酸性水溶液混合;经过晶化、过滤、干燥和焙烧后制得al-sba-15分子筛,编号为a-s-2,混合后的体系中p123三嵌段共聚物和无定形硅铝的质量比为2:1,所述的晶化温度为93℃,晶化时间为18h;晶化过程中ph控制为4.7,所述干燥温度控制在120℃,干燥时间为4h,所述焙烧温度控制在530℃,所述焙烧时间为5h。a-s-2分子筛性质见表1。a-s-2分子筛的xrd图谱与图1类似,显示出al-sba-15分子筛的特征峰。
68.(二)配置钼镍磷杂多酸溶液a,溶液的组成moo3的含量为45.5g/100ml,nio含量为7.5g/100ml,p的含量为3.5g/100ml,称取纳米氧化镁分散液8克(同实施例1),用去离子水稀释至100ml,得到溶液b;称取a-s-2分子筛6g,氧化铝干胶粉92g放入6l去离子水中搅拌溶解,使两种粉体均匀分散在体系中,得到浆液c,将溶液a和b并流加入到浆液c中,在超声波功率50hz条件下搅拌处理,温度48℃,处理40min后,得到混合浆液,然后经过过滤、洗涤,然后再在氮气保护下110℃下热处理4小时,得到含纳米氧化镁以及活性组分的第一催化剂前驱体;将第一催化剂前驱体粉碎后加入田菁粉4g,放入碾压机中,将柠檬酸4g,硝酸11.9g(浓度65%)溶解在132ml的去离子水里,制成酸性溶液,均匀倒入碾压机,经碾压25min后,物料呈膏状,在挤条机上挤成1.7mm三叶草,120℃干燥4小时,440℃焙烧3小时,得到第二催化剂前驱体;用含有核糖醇的水溶液浸渍第二催化剂前驱体,其中核糖醇的用量与mo原子摩尔比为1.5:1,浸渍后的样品室温晾干后,110℃热处理3h,160℃热处理1h,得到最终成品催化剂c-2,主要物理性质见表2和表3。
69.实施例3(一)al-sba-15分子筛及浆液的制备(1)无定形硅铝干胶a3的制备:铝酸钠溶液浓度30gal2o3/l,硅酸钠溶液浓度50gsio2/l,取0.75l铝酸钠溶液放入于成胶罐中,然后加入0.12l硅酸钠溶液,控制反应温度23℃,通入浓度为48v%的co2气体,通入co2气体占总通入量的50%时,一边通气一边加入0.20l硅酸钠溶液,控制成胶的ph值为8.8,然后通风稳定20分钟,浆液过滤并用75℃去离子水洗洗至中性,滤饼按固液体积比为11∶1加水打浆,在120℃,3.5mpa水蒸汽压力下处理2小时,在120℃干燥6小时后,粉碎过筛得无定形硅铝产品a3。将所制备的无定形硅铝a3和去离子水混合、打浆形成浆液;其中,无定形硅铝干胶与水的质量比为24:76;(2)配制含有p123三嵌段共聚物的酸性水溶液;将p123三嵌段共聚物加入到稀盐酸中,所述的稀盐酸溶液的浓度为0.16mol/l,含有p123三嵌段共聚物的酸性水溶液的ph为1.8,含有p123三嵌段共聚物的酸性水溶液的温度为33℃,所述含有p123三嵌段共聚物的酸性水溶液中p123三嵌段共聚物的含量为2.2wt%;(3)将步骤(1)制备的浆液同步骤(2)配制的含有p123三嵌段共聚物的酸性水溶液混合;经过晶化、过滤、干燥和焙烧后制得al-sba-15分子筛,编号为a-s-3,混合后的体系中p123三嵌段共聚物和无定形硅铝的质量比为2.5:1,所述的晶化温度为98℃,晶化时间为20h;晶化过程中ph控制为4.3,所述干燥温度控制在120 ℃,干燥时间为5h,所述焙烧温度控制在 540℃,所述焙烧时间为5h。a-s-3分子筛性质见表1。a-s-3分子筛的xrd图谱与图1类似,显示出al-sba-15分子筛的特征峰。
70.(二)配置钼镍磷杂多酸溶液a,溶液的组成moo3的含量为45.5g/100ml,nio含量为7.5g/100ml,p的含量为3.5g/100ml,称取纳米氧化镁分散液12克(杭州万景新材料有限公司生产的产品,分散液中氧化镁含量30wt%,粒径35nm),用去离子水稀释至100ml,得到溶液b;称取a-s-3分子筛10g,氧化铝干胶粉92g放入6l去离子水中搅拌溶解,使两种粉体均匀分散在体系中,得到浆液c,将溶液a和b并流加入到浆液c中,在超声波功率55hz条件下搅拌处理,处理温度为50℃,处理45min后,得到混合浆液,然后经过过滤、洗涤,然后再在氮气保护下120℃下热处理4小时,得到含纳米氧化镁以及活性组分的第一催化剂前驱体;将第一催化剂前驱体粉碎后加入田菁粉4g,放入碾压机中,将柠檬酸4g,硝酸12.5g(浓度65%)溶解在135ml的去离子水里,制成酸性溶液,均匀倒入碾压机,经碾压25min后,物料呈膏状,在挤条机上挤成1.7mm三叶草,120℃干燥4小时,450℃焙烧3小时,得到第二催化剂前驱体;用含有核糖醇的水溶液浸渍第二催化剂前驱体,其中核糖醇的用量与mo原子摩尔比为2:1,浸渍后的样品室温晾干后,120℃热处理3h,165℃热处理1.5h,得到最终成品催化剂c-3,主要物理性质见表2和表3。
71.实施例4(一)al-sba-15分子筛的制备其他条件同实施例1,只是在步骤(1)无定形硅铝干胶a1及浆液的制备过程中,控制成胶的ph值为9.8,得无定形硅铝产品a4,最终制备的分子筛a-s-4。a-s-4分子筛的xrd图谱与图1类似,显示出al-sba-15分子筛的特征峰。
72.(二)配置钼镍磷杂多酸溶液a,溶液的组成moo3的含量为45.5g/100ml,nio含量为7.5g/100ml,p的含量为3.5g/100ml,称取纳米氧化镁分散液15克(同实施例1),用去离子水稀释至100ml,得到溶液b;称取a-s-4分子筛12g,氧化铝干胶粉120g放入6l去离子水中搅拌溶解,使两种粉体均匀分散在体系中,得到浆液c,将溶液a和b并流加入到浆液c中,在超声波功率55hz条件下搅拌处理,处理温度为50℃,处理45min后,得到混合浆液,然后经过过滤、洗涤,然后再在氮气保护下120℃下热处理4小时,得到含纳米氧化镁以及活性组分的第一催化剂前驱体;将第一催化剂前驱体粉碎后加入田菁粉5g,将柠檬酸4g,硝酸12.8g(浓度65%)溶解在138ml的去离子水里,制成酸性溶液,均匀倒入碾压机,经碾压25min后,物料呈膏状,在挤条机上挤成1.7mm三叶草,110℃干燥4小时,450℃焙烧3小时,得到第二催化剂前驱体,用含有d-甘露糖醇的水溶液浸渍第二催化剂前驱体,其中d-甘露糖醇的用量与mo原子摩尔比为0.8:1,浸渍后的样品室温晾干后,120℃热处理3h,175℃热处理1.8h,得到最终成品催化剂c-4,主要物理性质见表2和表3。
73.实施例5(一)al-sba-15分子筛的制备(1)无定形硅铝干胶a5及浆液的制备:铝酸钠溶液浓度20gal2o3/l,硅酸钠溶液浓度50gsio2/l,取0.75l铝酸钠溶液放入于成胶罐中,然后加入0.12l硅酸钠溶液,控制反应温度23℃,通入浓度为45v%的co2气体,控制成胶的ph值为8.8停止,然后通风稳定20分钟,浆液过滤并用75℃去离子水洗洗至中性,滤饼按固液体积比为11∶1加水打浆,在120℃,3.5mpa水蒸汽压力下处理2小时,在120℃干燥6小时后,粉碎过筛得无定形硅铝产品a5。将所制备的无定形硅铝a5和去离子水混合、打浆形成浆液;其中,无定形硅铝干胶与水的质量比为24:76;
(2)配制含有p123三嵌段共聚物的酸性水溶液;将p123三嵌段共聚物加入到稀盐酸中,所述的稀盐酸溶液的浓度为0.16mol/l,含有p123三嵌段共聚物的酸性水溶液的ph为1.8,含有p123三嵌段共聚物的酸性水溶液的温度为33℃,所述含有p123三嵌段共聚物的酸性水溶液中p123三嵌段共聚物的含量为2.8wt%;(3)将步骤(1)制备的浆液同步骤(2)配制的含有p123三嵌段共聚物的酸性水溶液混合;经过晶化、过滤、干燥和焙烧后制得al-sba-15分子筛,编号为a-s-5,混合后的体系中p123三嵌段共聚物和无定形硅铝的质量比为2.5:1,所述的晶化温度为98℃,晶化时间为20h;晶化过程中ph控制为4.3,所述干燥温度控制在120℃,干燥时间为5h,所述焙烧温度控制在 540℃,所述焙烧时间为5h。a-s-5分子筛性质见表1。a-s-5分子筛的xrd图谱与图1类似,显示出al-sba-15分子筛的特征峰。
74.(二)配置钼镍磷杂多酸溶液a,溶液的组成moo3的含量为45.5g/100ml,nio含量为7.5g/100ml,p的含量为3.5g/100ml,称取纳米氧化镁分散液8克(同实施例1),用去离子水稀释至100ml,得到溶液b;称取a-s-2分子筛15g,氧化铝干胶粉125g放入8l去离子水中搅拌溶解,使两种粉体均匀分散在体系中,得到浆液c,将溶液a和b并流加入到浆液c中,在超声波功率53hz条件下搅拌处理,处理温度为42℃,处理40min后,得到混合浆液,然后经过过滤、洗涤,然后再在氮气保护下120℃下热处理4小时,得到含纳米氧化镁以及活性组分的第一催化剂前驱体;将第一催化剂前驱体粉碎后加入田菁粉5g,放入碾压机中,将柠檬酸5g,硝酸13.5g(浓度65%)溶解在141ml的去离子水里,制成酸性溶液,均匀倒入碾压机,经碾压25min后,物料呈膏状,在挤条机上挤成1.7mm三叶草,120℃干燥4小时,430℃焙烧3小时,得到第二催化剂前驱体,用含有d-甘露糖醇和水苏糖的水溶液浸渍第二催化剂前驱体,其中d-甘露糖醇的用量与mo原子摩尔比为0.5:1,水苏糖的用量与mo原子摩尔比为0.8:1,浸渍后的样品室温晾干后,130℃热处理3h,155℃热处理1.5h,得到最终成品催化剂c-5,主要物理性质见表2和表3。
75.对比例1(1)页岩油加氢处理催化剂载体的制备取大孔氢氧化铝干胶粉120g,小孔氧化铝干胶粉50g,加入柠檬酸和田菁粉各5g,混合均匀。然后均匀加入稀硝酸水溶液155g,其中硝酸质量为12.5g(浓度65%)。将物料混捏25min,然后碾压25min,用直径1.7mm的三叶草孔板挤条。经120℃干燥4h后550℃焙烧4h。焙烧后的载体记为z-1。
76.(2)催化剂制备用含mo、ni、p的浸渍液等体积浸渍载体z-1,室温晾干后,经110℃干燥4h,435℃焙烧3h后,最终获得的催化剂记为c-6。催化剂性质见表2和表3。
77.对比例2分别称取模板剂三嵌共聚物p123和硅源正硅酸乙酯,其中模板剂p123质量为5.5g,正硅酸乙酯质量为10.2g;将模板剂和硅源加入ph为2.8的hcl溶液中,28℃下充分搅拌30h;将搅拌后的混合物120℃静置晶化20h,去离子水洗涤,干燥后得到sba-15。将得到的sba-15分子筛进行打浆,固液比为1:10,然后加入含有23g异丙醇铝的盐酸溶液中,升温至100℃,搅拌20h,过滤、洗涤后60℃干燥过夜,550℃焙烧5h,得到介孔材料a-s-8,性质见表1。
78.催化剂的制备方法同实施例1,不同之处把a-s-1改为a-s-8催化剂编号c-7。催化
剂性质见表2和表3。
79.对比例3取6.2gp123加入到600ml0.18mol/l的盐酸溶液中,升温到26℃后恒温搅拌6小时,p123完全溶解后,溶液呈透明状态。加入5.2gy分子筛浆液,ph值控制在3.3,恒温搅拌反应6小时,升温至98℃水热晶化24小时。然后,过滤、洗涤,在120℃干燥6小时,550℃焙烧6小时,得到al-sba-15介孔分子筛,编号为a-s-9,性质见表1。
80.催化剂的制备方法同实施例1,不同之处把a-s-1改为a-s-9催化剂编号c-8。催化剂性质见表2和表3。
81.对比例4将高岭土在700℃下焙烧活化4h,称取焙烧后高岭土12g,采用6mol/l盐酸浸处理4h,之后去离子水抽滤洗涤至中性并烘干;将烘干后的样品在900℃下焙烧2h;之后放入5mol/l的naoh碱溶液中,高温高压下反应3h(温度为160℃,压力为0.5mpa),反应完成后,调节其ph值在14.0。之后将其逐滴加入到表面活性剂和酸的混合溶液中(n(fso-100)/n(p123)=5.5),盐酸浓度为7.5mol/l,40℃搅拌反应24h,160℃下水热反应48h,过滤、洗涤、干燥后在马弗炉550℃下焙烧6h,得到介孔材料a-s-10,性质见表1。
82.催化剂的制备方法同实施例1,不同之处把a-s-1改为a-s-10,催化剂编号c-9。催化剂性质见表2和表3。
83.对比例5将4gp123加入到2mol/l125ml的盐酸溶液中,40℃下搅拌,直至p123完全溶解;将8.5g正硅酸乙酯加入到含有p123的盐酸溶液中,搅拌4h,加入硝酸铝使硅铝摩尔比为35,继续搅拌20h,将上述溶液加入到250ml反应釜内,100℃条件下搅拌48h,冷却到室温后,用氨水溶液调整ph值到7.5,不断搅拌条件下,升温至100℃,搅拌72h,过滤、洗涤后60℃干燥过夜,550℃焙烧6h,得到介孔材料a-s-11,性质见表1。
84.催化剂的制备方法同实施例1,不同之处把a-s-1改为a-s-11,催化剂编号c-10。催化剂性质见表2和表3。
85.表1 al-sba-15分子筛性质项目a-s-1a-s-2a-s-3a-s-4a-s-5比表面积,m2/g735737745750748氧化铝含量,wt%31.2539.0658.4431.2571.40孔容,ml/g1.171.131.091.151.13中强酸酸量,ml/g0.760.770.820.850.83b/l0.3170.2610.2450.3210.332孔分布,%
ꢀꢀꢀꢀꢀ
<4nm11.1513.6312.8314.2514.894~15nm54.6253.6553.0255.5658.35>15nm34.2332.7234.1530.1926.76续表1项目a-s-8a-s-9a-s-10a-s-11比表面积,m2/g706720695708
氧化铝含量,wt%17.254813孔容,ml/g1.040.850.781.05中强酸酸量,ml/g0.450.530.410.43b/l1.251.211.241.32孔分布,%
ꢀꢀꢀꢀ
<4nm43.0542.6946.2845.364~15nm37.5638.2535.6936.45>15nm19.3919.0618.0318.19表2 催化剂的组成和性质催化剂编号c-1c-2c-3c-4c-5组成
ꢀꢀꢀꢀꢀ
moo3,wt%23.3023.8023.5023.623.8nio,wt%4.23.93.84.64.2p,wt%1.61.71.81.61.3纳米氧化镁,wt%2.433.64.55.4al-sba-15分子筛,wt%6.24.77.68.69.4性质
ꢀꢀꢀꢀꢀ
比表面积,m2/g223225228226228孔容,ml/g0.360.370.380.350.34酸分布
ꢀꢀꢀꢀꢀ
弱酸,%31.3735.7537.5834.5833.65中强酸,%58.6255.2354.3056.8957.26强酸,%10.019.028.128.539.09分散度
ꢀꢀꢀꢀꢀ
mo/al0.1720.1780.1800.1760.178ni/al0.070.080.070.080.07续表2催化剂编号c-6c-7c-8c-9c-10组成
ꢀꢀꢀꢀꢀ
moo3,wt%23.9123.3023.3023.3023.30nio,wt%3.934.24.24.24.2p,wt%1.491.61.61.61.6性质
ꢀꢀꢀꢀꢀ
比表面积,m2/g186212215209218孔容,ml/g0.300.310.320.330.33酸分布
ꢀꢀꢀꢀꢀ
弱酸,%33.1237.4437.6236.5735.62中强酸,%38.2048.3646.0247.3848.03强酸,%28.6814.2016.3616.0516.35
分散度
ꢀꢀꢀꢀꢀ
mo/al0.1250.1530.1470.1510.148ni/al0.050.040.050.070.06表3 运转2000小时催化剂强度对比
催化剂编号c-1c-2c-3c-4c-5c-6新剂压碎强度,n
·
cm-1
162163165162163165运转后催化剂压碎强度,n
·
cm-1
162162165162164121
催化剂评价实施例和对比例的催化剂活性评价实验在100ml小型固定床加氢装置上进行,评价前对催化剂进行预硫化。催化剂评价条件为在反应总压14.5mpa,液时体积空速0.2h-1
,氢油体积比1200:1,反应温度为383℃。活性评价实验用原料油性质见表4,活性评价结果见表5。
86.表4 原料油性质原料油名称页岩油全馏分密度(20℃)/g
·
cm-3
0.920馏程/℃ ibp/10%167/24830%/50%316/37470%/90%428/50195%/ebp-/663粘度(50℃)/mm2·
s-1
3.471粘度(100℃)/mm2·
s-1
11.70凝点/℃35闪点/℃(闭口) 残炭,wt%2.47酸值/(mgkoh)
·
g-1
0.48沥青质,wt%0.08s,wt%0.53n,wt%1.09c,wt%84.17h,wt%11.35o,wt%0.85质谱组成,wt% 链烷烃19.3总环烷27.1总芳烃32.7总胶质20.9表5 活性评价结果催化剂编号氮含量,μg
·
g-1
c-110.3
c-29.9c-39.8c-410.2c-59.7c-643.5c-722.3c-825.3c-923.5c-1021.5由表5可见,与对比例催化剂相比,采用本发明制备的页岩油加氢处理催化剂加氢脱氮活性显著提高,抗水性能力增强。
87.表6 无定形硅铝的性质无定形硅铝编号a1a2a3a4a5比表面积,m2/g512537528535519孔容,ml/g1.181.231.201.261.19孔分布,%
ꢀꢀꢀꢀꢀ
4~15nm8886879293>15nm34332
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1