一种复合纳滤膜及其制备方法和应用
1.本发明属于纳滤膜技术领域,具体涉及一种复合纳滤膜及其制备方法和应用。
背景技术:
2.随着纺织印染行业的快速发展,每年产生和排放大量的纺织废水。它通常由无机盐、染料等化学物质组成,如果不进行适当的处理,这些物质对环境会产生危害。因此,从纺织废水中分离染料和盐类物质是防止废水污染生态环境的关键。
3.纳滤膜是一种孔径结构一致,且孔径范围为1~2nm的微孔过滤膜。纳滤膜可以有效分离直径为200~1000da之间的污染物。目前,纳滤膜常用于处理工业废水(如用于染料废水中染料的清除),海水淡化等领域。膜分离在污水处理中起着至关重要的作用,因其操作简单、能耗低、维护费用低等优点被公认为一种先进的分离技术。与其他类型的膜相比,纳滤膜对多价离子和小分子有机物具有良好的截留能力,在纺织废水的淡化和回收中得到了较好的利用。但是纳滤膜作为一种压力驱动膜,在水处理中会不可避免的出现膜污染问题。在膜运行过程中,污染物质(如多价离子或染料)很容易吸附在膜表面及膜孔内部,使膜孔不可避免地造成堵塞,致使纯水通量及脱除率下降,降低膜的运行寿命,同时增加膜的使用成本。
技术实现要素:
4.有鉴于此,本发明提供了一种复合纳滤膜及其制备方法和应用,本发明提供的复合纳滤膜具有较高的纯水通量和耐污染性能,延长了复合纳滤膜的使用寿命。
5.为了解决上述技术问题,本发明提供了一种复合纳滤膜,包括依次层叠的基底层、基膜层和聚酰胺分离层;
6.所述基膜层包括聚砜膜和分散在所述聚砜膜中的氧化二硫化钼;
7.所述聚酰胺分离层由水相溶液和油相溶液通过原位界面聚合反应制备得到;所述水相溶液中含有哌嗪,所述油相溶液中含有均苯三甲酰氯。
8.优选的,所述水相溶液包括以下质量百分含量的组分:
[0009][0010]
所述油相溶液为均苯三甲酰氯的正己烷溶液,所述均苯三甲酰氯的质量在正己烷中的浓度为0.1~0.5g:1/100ml。
[0011]
优选的,所述氧化二硫化钼的粒径为10~2000nm,所述氧化二硫化钼的水接触角为20~45
°
。
[0012]
本发明还提供了上述技术方案所述复合纳滤膜的制备方法,包括以下步骤:
[0013]
将有机溶剂、有机致孔剂、氧化二硫化钼和聚砜混合,得到铸膜液;
[0014]
将所述铸膜液脱泡后,在基底层表面成膜,成膜后进行浸泡处理得到基膜层,得到初级复合纳滤膜;
[0015]
将所述初级复合纳滤膜依次于水相溶液和油相溶液中浸泡,进行原位界面聚合反应,得到所述复合纳滤膜;所述水相溶液中含有哌嗪,所述油相溶液中含有均苯三甲酰氯。
[0016]
优选的,所述脱泡为在恒温和真空的条件下静置,所述脱泡的温度为25~70℃,所述真空的真空度为0.3~0.8mpa,所述脱泡的时间为1~8h。
[0017]
优选的,所述成膜包括刮膜、空气浴和凝胶浴;所述刮膜的速度为2~5m/min,所述刮膜的厚度为50~200μm;
[0018]
所述空气浴的温度为25~80℃,时间为10~240s;
[0019]
所述凝胶浴的温度为15~40℃,时间为4~48h。
[0020]
优选的,在水相溶液中的浸泡时间为10~30s,在油相溶液中的浸泡时间为10~30;
[0021]
所述原位界面聚合反应的温度为50~70℃,时间为1~5min。
[0022]
优选的,所述有机溶剂包括n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺和n
‑
甲基吡咯烷酮中的一种或多种。
[0023]
优选的,所述有机致孔剂包括聚乙二醇、丙三醇、丙二醇和丙酮中的一种或多种。
[0024]
本发明还提供了上述技术方案所述复合纳滤膜或上述技术方案所述的制备方法制备得到的复合纳滤膜在水处理、染料浓缩或海水淡化中的应用。
[0025]
本发明提供了一种复合纳滤膜,包括依次层叠的基底层、基膜层和聚酰胺分离层;所述基膜层包括聚砜膜和分散在所述聚砜膜中的氧化二硫化钼;所述聚酰胺分离层由水相溶液和油相溶液通过原位界面聚合反应制备得到;所述水相溶液中含有哌嗪,所述油相溶液中含有均苯三甲酰氯。本发明提供的复合纳滤膜的基膜层中含有的氧化二硫化钼,不仅有效调节了复合纳滤膜表面的亲水性,增加了水通过复合纳滤膜的效率,而且氧化二硫化钼使复合纳滤膜表面具有更强的负电性,使复合纳滤膜表面和污染物之间产生静电排斥作用,减少污染物在复合纳滤膜表面的聚集,从而提高复合纳滤膜表面的抗污染性;氧化二硫化钼具有层状结构,氧化二硫化钼的分子层间存在孔隙,能够起到水通道的作用,使水分子快速通过,从而进一步提高复合纳滤膜纯水通量。本发明通过原位界面聚合反应得到聚酰胺分离层具有较高的致密性,进而提高了复合纳滤膜对污染物(废水中多价离子和染料)的脱除率。本发明中氧化二硫化钼与聚酰胺之间形成氢键从而从而致密皮层进一步提高了复合纳滤膜对污染物的脱除率。由实施例结果可知,在0.4mpa操作压力下,本发明提供的psf超滤基膜中氧化二硫化钼含量为0.06wt.%时改性复合纳滤膜对染料孟加拉玫瑰红的脱除率为99.8%,对na2so4的脱除率为95.3%,纯水通量为27.7lm
‑2h
‑1bar
‑1。
附图说明
[0026]
图1为实施例1和对比例制备得到的复合纳滤膜的结构示意图;
[0027]
图2为实施例2和对比例1制备得到的复合纳滤膜的膜孔径分布图;
[0028]
图3为实施例2和对比例1制备得到的复合纳滤膜的平面和断面的sem图;
[0029]
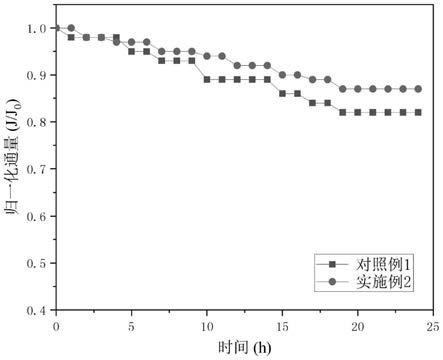
图4为实施例2和对比例1制备得到的复合纳滤膜长期运行归一化通量对比曲线
图。
具体实施方式
[0030]
本发明提供了一种复合纳滤膜,包括依次层叠的基底层、基膜层和聚酰胺分离层;
[0031]
所述基膜层包括聚砜膜和分散在所述聚砜膜中的氧化二硫化钼;
[0032]
所述聚酰胺分离层由水相溶液和油相溶液通过原位界面聚合反应制备得到;所述水相溶液中含有哌嗪,所述油相溶液中含有均苯三甲酰氯。
[0033]
在本发明中,若无特殊说明,所有原料组分均为本领域技术人员熟知的市售产品。
[0034]
在本发明中,所述复合纳滤膜包括基底层;所述基底层优选包括无纺布。在本发明中,所述无纺布的厚度优选为75~150μm,更优选为80~100μm,更进一步优选为97μm;所述无纺布的密度优选为0.73~0.85g/m3,更优选为0.77g/m3。在本发明中,所述无纺布对基膜层起支撑作用。
[0035]
在本发明中,所述复合纳滤膜包括基膜层;所述基膜层的厚度优选为106~118μm,更优选为110~115μm。在本发明中,所述基膜层优选由包括以下质量份数组分的铸膜液制膜得到:
[0036][0037]
在本发明中,以质量份数计,制备所述基膜层用铸膜液的原料包括15~30份聚砜(psf),优选为20~25份。在本发明中,所述聚砜作为基体膜材料,提高了复合纳滤膜的机械性能。
[0038]
以所述聚砜的质量份数为基准,制备所述基膜层用铸膜液的原料包括64~90.1份有机溶剂,优选为70~85份,更优选为75~81份。在本发明中,所述有机溶剂优选包括n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺和n
‑
甲基吡咯烷酮中的一种或多种,更优选为n,n
‑
二甲基乙酰胺;当所述有机溶剂为上述具体物质中的多种时,本发明对所述具体物质的配比没有特殊限定。
[0039]
以所述聚砜的质量份数为基准,制备所述铸膜液的原料包括0.01~6份有机致孔剂,优选为1~5份。在本发明中,所述有机致孔剂优选包括聚乙二醇、丙三醇、丙二醇和丙酮中的一种或多种,更优选为聚乙二醇,所述聚乙二醇优选为聚乙二醇400;当所述有机致孔剂为上述具体选择中的两种以上时,本发明对所述具体物质的配比无特殊限定,采用任意配比即可。
[0040]
在本发明中,所述有机致孔剂能够提高铸膜液的粘度,从而提高复合纳滤膜的致密性,进而提高复合纳滤膜的脱除率。
[0041]
以所述聚砜的质量份数为基准,制备所述基膜层用铸膜液的原料包括0.01~3份氧化二硫化钼,优选为0.03~0.5份,更优选为0.06~0.12份。在本发明中,所述氧化二硫化钼的粒径优选为10~2000nm,更优选为100~800nm,所述氧化二硫化钼的水接触角优选为20~45
°
,更优选为30~40
°
。
[0042]
在本发明中,所述氧化二硫化钼优选通过hummers氧化法制备得到。
[0043]
所述氧化二硫化钼的制备方法,优选包括以下步骤:
[0044]
将二硫化钼和硝酸钠进行第一混合,得到混合物;
[0045]
将浓硫酸和所述混合物进行第二混合,得到分散液;
[0046]
将所述分散液和高锰酸钾进行第三混合,发生氧化反应,得到氧化二硫化钼分散液;
[0047]
将所述氧化二硫化钼分散液进行除杂、过滤和干燥,得到氧化二硫化钼。
[0048]
本发明将二硫化钼和硝酸钠进行第一混合,得到混合物。在本发明中,所述二硫化钼和硝酸钠的质量比优选为2.8~3.2:1,更优选为3:1。本发明对所述第一混合无特殊限定,采用本领域技术人员熟知的过程进行并保证所述二硫化钼和硝酸钠充分混合均匀即可。
[0049]
得到混合物后,本发明将浓硫酸和所述混合物进行第二混合,得到分散液。在本发明中,所述浓硫酸的质量浓度优选为98%;所述二硫化钼的质量和所述浓硫酸的体积比优选为2.8~3.2g:50ml,更优选为3g:50ml;所述第二混合的方式优选为搅拌,所述搅拌的转速优选为430~470r/min,更优选为450r/min,时间优选为11~13h,更优选为12h。
[0050]
得到分散液后,将所述分散液和高锰酸钾进行第三混合,发生氧化反应,得到氧化二硫化钼分散液。在本发明中,所述高锰酸钾和所述二硫化钼的质量比优选为1.8~2.2:1,更优选为2:1。本发明对所述第三混合无特殊限定,采用本领域技术人员熟知的过程,保证所述高锰酸钾充分分散在所述分散液中即可。
[0051]
在本发明中,所述氧化反应优选包括两步氧化反应,第一步氧化反应的温度优选为0~5℃,更优选为0~1℃,所述第一步氧化反应的时间优选为0.2~1h,更优选为0.5~0.6h;第二步氧化反应的温度优选为33~37℃,更优选为35℃,时间优选为2.8~3.2h,更优选为3h。在本发明中,所述第一步氧化反应优选在冰浴中进行;所述第二步氧化反应优选在油浴中进行,所述油浴的过程优选在搅拌的条件下进行,本发明对所述搅拌无特殊限定,采用本领域技术人员熟知的过程进行即可。
[0052]
得到氧化二硫化钼分散液后,本发明将所述氧化二硫化钼分散液进行除杂、过滤、干燥,得到氧化二硫化钼。在本发明中,所述除杂优选为:将所述氧化二硫化钼分散液进行冰浴后,依次加入双氧水和盐酸;本发明在所述冰浴过程中和冰浴结束后优选加入去离子水,所述去离子水的体积与所述二硫化钼的质量比优选为148~152ml:3g,更优选为150ml:3g。在所述冰浴的过程中,本发明优选加入去离子水总量的30~35%,添加去离子水的目的是稀释浓硫酸;冰浴结束后加入剩余的去离子水,控制氧化二硫化钼分散液温度在60℃以下。在本发明中,所述冰浴的过程优选在搅拌的条件下进行,本发明对所述搅拌无特殊限定,采用本领域技术人员熟知的过程达到搅拌均匀的目的即可。
[0053]
在本发明中,所述双氧水的质量浓度优选为28~32%,更优选为30%;所述双氧水的体积与所述二硫化钼的质量比优选为7.8~8.2ml:3g,更优选为8ml:3g。在本发明中,加入去离子水和双氧水的目的是可以更有效的去除所述产物体系中过剩的高锰酸钾。在本发明中,所述盐酸的质量浓度优选为0.08~1.2%,更优选为0.1%;所述盐酸的体积和所述二硫化钼的质量比优选为248~252ml:3g,更优选为250ml:3g。在本发明中,加入所述盐酸的目的是去除所述产物体系中的金属元素。
[0054]
本发明对所述过滤和干燥均没有任何特殊的限定,采用本领域技术人员熟知的过程进行即可。
[0055]
在本发明中,所述氧化二硫化钼的粒径优选为10~2000nm,更优选为100~800nm,所述氧化二硫化钼的水接触角优选为20~45
°
,更优选为30~40
°
。在本发明中,所述氧化二硫化钼具有优良亲水性、电负性及机械性能,氧化二硫化钼的添加能够提高复合纳滤膜的致密性,进而提高复合纳滤膜脱除率;同时氧化二硫化钼的加入使纳滤膜表面具有更强的亲水性,荷负电性,从而提高了基膜改性复合纳滤膜的纯水通量;此外,氧化二硫化钼的分子间存在孔隙,能够起到水通道的作用,使水分子快速通过,从而进一步提高纳滤膜纯水通量。
[0056]
在本发明中,所述基膜层的孔隙率优选为70~90%,更优选为75~85%。
[0057]
在本发明中,所述复合纳滤膜还包括聚酰胺分离层;所述基膜层的厚度优选为80~120nm,更优选为100~110nm。在本发明中,所述聚酰胺分离层由水相溶液和油相溶液通过原位界面聚合反应制备得到。在本发明中,所述水相溶液优选包括以下质量百分含量的组分:
[0058][0059]
以质量百分含量计,所述水相溶液优选包括1~2%哌嗪(pip),更优选为1.3~1.6%。
[0060]
以质量百分含量计,所述水相溶液优选包括1.5~3.0%ph值调节剂,更优选为2.0~2.5%。在本发明中,所述水相溶液的ph值优选为9.8~10.2,更优选为10。本发明对所述ph值调节剂的种类和用量无特殊限定,只要能够使水相溶液的ph达到要求即可。在本发明的实施例中,所述ph值调节剂为樟脑磺酸和三乙胺的混合物,所述樟脑磺酸和三乙胺的质量比为1:1。
[0061]
以质量百分含量计,所述水相溶液优选包括0.58~0.62%十二烷基磺酸钠,更优选为0.6%。在本发明中,所述十二烷基磺酸钠能够降低基膜表面张力。
[0062]
以质量百分含量计,所述水相溶液优选还包括余量的水。在本发明中,所述水优选为去离子水。
[0063]
在本发明中,所述油相溶液优选为均苯三甲酰氯(tmc)的正己烷溶液,所述均苯三甲酰氯在正己烷中的浓度优选为0.1~0.5g/100ml,更优选为0.2~0.35g/100ml。
[0064]
在本发明中,所述原位界面聚合反应的温度优选为50~70℃,更优选为55~65℃;所述原位界面聚合反应的时间优选为1~5min,更优选为2~4min。
[0065]
在本发明中,所述界面反应优选为哌嗪和均苯三甲酰氯反应生成聚酰胺,反应方程式如式1所示:
[0066][0067]
在本发明中,所述聚酰胺包括两种组分,一种为全交联聚哌嗪酰胺,一种为包括网络交联部分和线性交联部分等聚哌嗪酰胺;本发明对所述两种组分的含量无特殊限定。
[0068]
本发明限定水相溶液的ph值为9.8~10.2,可以中和界面反应生成的hcl,促进界面聚合反应向正反应方向进行。
[0069]
本发明还提供了上述技术方案所述复合纳滤膜的制备方法,包括以下步骤:
[0070]
将有机溶剂、有机致孔剂、氧化二硫化钼和聚砜混合,得到铸膜液;
[0071]
将所述铸膜液脱泡后,在基底层表面成膜,成膜后进行浸泡处理得到基膜层,得到初级复合纳滤膜;
[0072]
将所述初级复合纳滤膜依次于水相溶液和油相溶液中浸泡,进行原位界面聚合反应,得到所述复合纳滤膜;所述水相溶液中含有哌嗪,所述油相溶液中含有均苯三甲酰氯。
[0073]
本发明将有机溶剂、有机致孔剂、氧化二硫化钼和聚砜混合,得到铸膜液。在本发明中,所述混合优选包括以下步骤:
[0074]
将有机溶剂和有机致孔剂进行第四混合,得到致孔剂溶液;
[0075]
将所述致孔剂溶液和氧化二硫化钼进行第五混合,得到混合分散液;
[0076]
将所述混合分散液和聚砜进行第六混合,得到铸膜液。
[0077]
本发明将有机溶剂和有机致孔剂进行第四混合,得到致孔剂溶液。在本发明中,所述第四混合优选在搅拌的条件下进行,所述搅拌的转速优选为300~600r/min,更优选为400~500r/min,更进一步优选为450r/min;所述搅拌的时间优选为1~5h,更优选为2~3h。在本发明中,所述有机溶剂和有机致孔剂经过所述第四混合,能够使有机致孔剂与有机溶剂均匀混合,保证有机致孔剂在铸膜液中均匀分布,从而使基膜具有分布更加均匀的膜孔。
[0078]
得到所述致孔剂溶液后,本发明将所述致孔剂溶液和氧化二硫化钼进行第五混合,得到混合分散液。在本发明中,所述第五混合优选包括依次进行的超声和搅拌;所述超声的功率优选为500~10000w,更优选为500~2000w,所述超声的时间优选为2~36h,更优选为4~12h;所述搅拌的转速优选为100~600r/min,更优选为300~500r/min,更进一步优选为400r/min;所述搅拌的时间优选为0.5~4h,更优选为1~2h。在本发明中,所述超声和搅拌可以使氧化二硫化钼更均匀分散于分散液中。
[0079]
得到所述混合分散液后,本发明将所述混合分散液和聚砜进行第六混合,得到铸膜液。在本发明中,所述第六混合优选在搅拌的条件下进行,所述搅拌的转速优选为50~200r/min,更优选为100~150r/min;所述搅拌的时间优选为0.5~5h,更优选为1~2h。在本发明中,所述搅拌使聚砜充分溶解。
[0080]
本发明采用分步混合的方式能够使各组分混合均匀,同时避免各组分间以及各组分和氧化二硫化钼间发生团聚。
[0081]
得到所述铸膜液后,本发明将所述铸膜液脱泡后,在基底层表面成膜,成膜后进行浸泡处理得到基膜层,得到初级复合纳滤膜。在本发明中,所述脱泡优选为在恒温和真空的条件下静置,所述脱泡的温度优选为25~80℃,更优选为50℃;所述真空的真空度优选为0.2~0.9mpa,更优选为0.5~0.85mpa,更进一步优选为0.8mpa;所述脱泡的时间优选为1~12h,更优选为3~10h,更进一步优选为4~6h。在本发明中,所述脱泡能够除去铸膜液中的气泡防止基膜改性复合纳滤膜中产生大空腔,更进一步的降低脱除率。
[0082]
在本发明中,所述成膜优选包括刮膜、空气浴和凝胶浴。在本发明中,所述刮膜优选采用刮刀在基底层表面进行刮膜,所述刮膜的厚度优选为20~300μm,更优选为30~150μm,更进一步优选为50~100μm。在本发明中,所述刮膜的环境温度优选为24~26℃,更优选为25℃,所述刮膜的环境相对湿度优选为30~80%,更优选为30~50%。在本发明中,所述刮膜的速度优选为1~5m/min,更优选为1.5~3m/min,所述刮刀优选含有凹槽,所述凹槽的深度优选为50~350μm,可以具体为50μm、100μm、150μm、200μm、250μm、300μm。在本发明中,所述刮膜后优选还包括:将刮膜得到的产物在温度为70~80℃的空气中蒸发0.45~0.55min后在水中固化。在本发明中,所述水优选为自来水,所述水的温度优选为常温,更优选为23~25℃;所述固化的时间优选为0.4~0.6min,更优选为0.5min。
[0083]
在本发明中,所述空气浴的温度优选为25~90℃,更优选为60~90℃,更进一步优选为80℃,时间优选为5~320s,更优选为20~60s,更进一步优选为30s。所述凝胶浴的温度优选为15~50℃,更优选为20~30℃,更进一步优选为25℃,时间优选为0.1~48h,更优选为0.5~24h,更进一步优选为10~12h。所述凝胶浴优选包括自来水、乙醇、丙酮和二甲基乙酰胺中的一种或多种,更优选包括自来水、二甲基乙酰胺、乙醇和丙酮的混合液或自来水。在本发明中,当所述凝胶浴包括两种以上上述具体物质时,本发明对具体物质之间的配比无特殊限定,采用任意配比即可。
[0084]
在本发明中,所述浸泡处理优选包括以下步骤:将所述成膜后产物依次在纯水和质量浓度为30%的甘油水溶液中浸泡后取出并晾干,得到所述初级复合纳滤膜。在本发明中,在纯水中进行浸泡的时间优选为47~49h,更优选为48h,在甘油水溶液中进行浸泡的时间优选为12~48h,更优选为24h,所述浸泡的温度优选为24~28℃,更优选为25℃。本发明将所述成膜后产物在纯水中浸泡的目的是使有机致孔剂溶于纯水中,保证初级复合纳滤膜中形成膜孔;本发明将所述成膜后产物在质量浓度为30%的甘油水溶液中浸泡的目的是防止基膜中膜孔收缩导致纯水通量下降。
[0085]
得到初级复合纳滤膜后,本发明将所述初级复合纳滤膜依次于水相溶液和油相溶液中浸泡,进行原位界面聚合反应,得到所述复合纳滤膜。本发明在进行浸泡前优选还包括:将所述初级复合纳滤膜在水中浸泡后冲洗、干燥。在本发明中,所述水优选为去离子水;所述浸泡的时间优选为1~6h,更优选为2~5h。在本发明中,所述冲洗用溶剂优选为去离子
水。在本发明中,所述干燥优选为晾干。本发明通过浸泡和冲洗能够除去纳滤膜表面残留的聚乙二醇。
[0086]
在本发明中,以质量百分含量计,所述水相溶液优选包括1~2%哌嗪(pip),更优选为1.3~1.6%;优选包括1.5~3.0%樟脑磺酸,更优选为2.0~2.5%;优选包括1.5~3.0%三乙胺,更优选为2.0~2.5%;优选包括0.58~0.62%十二烷基磺酸钠,更优选为0.6%;还优选包括余量的水。在本发明中,所述水优选为去离子水。本发明将所述初级复合纳滤膜在水相溶液中的浸泡时间优选为10~30s,更优选为20~30s。本发明对所述水相溶液的用量无特殊限定,只要能够浸没所述初级复合纳滤膜即可。本发明将所述初级复合纳滤膜在水相溶液中浸泡完成取出后优选还包括:将浸泡完成的初级复合纳滤膜表面多余的水相溶液除去。在本发明中,除去初级复合纳滤膜表面多余水相溶液的方式优选包括以下三种方式:第一种方式为将多余水相溶液吹干;第二种方式为自然晾干;第三种方式为利用纸巾吸附多余水相溶液。
[0087]
在本发明中,所述油相溶液优选为均苯三甲酰氯(tmc)的正己烷溶液,所述均苯三甲酰氯在正己烷中的浓度优选为0.1~0.5g/100ml,更优选为0.2~0.35g/100ml。本发明将浸泡水相溶液的初级复合纳滤膜在油相溶液中的浸泡时间优选为10~30s,更优选为15~20s。本发明对所述油相溶液的用量无特殊限定,只要能够浸没初级复合纳滤膜即可。本发明将所述初级复合纳滤膜在油相溶液中浸泡完成取出后优选还包括:将浸泡完成的初级复合纳滤膜表面多余的油相溶液除去。本发明除去多余的油相溶液的方式与除去多余水相溶液的方式一致,在此不再重复赘述。
[0088]
在本发明中,所述原位界面聚合反应的温度优选为50~70℃,更优选为55~65℃;时间优选为1~5min,更优选为2~3min。
[0089]
得到复合纳滤膜后本发明优选将复合纳滤膜保存在去离子水中。
[0090]
本发明通过控制水相和油相的浓度和反应时间来控制聚酰胺分离层的形成速率从而提高聚酰胺分离层的强度,在本发明中聚酰胺形成速率越快越容易形成致密皮层,但是太快又会产生皮层缺陷。
[0091]
本发明还提供了上述技术方案所述复合纳滤膜或上述技术方案所述的制备方法制备得到的复合纳滤膜在水处理、染料浓缩或海水淡化中的应用。本发明对所述应用的方式无特殊要求,采用本领域常规的方式即可。
[0092]
为了进一步说明本发明,下面结合实施例对本发明提供的技术方案进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
[0093]
实施例1
[0094]
制备氧化二硫化钼:
[0095]
将3g二硫化钼和1g硝酸钠混合,得到混合物;
[0096]
将50ml质量浓度为98%的浓硫酸和所述混合物混合,按照450r/min的转速搅拌12h,得到分散液;
[0097]
在冰浴条件下,向分散液中加入6g高锰酸钾,进行第一步氧化反应,反应30min时间后在35℃的油浴中进行第二步氧化反应,反应3h后,得到氧化二硫化钼分散液;其中油浴过程中伴随搅拌。
[0098]
将所述氧化二硫化钼分散液依次进行除杂、过滤和干燥,得到氧化二硫化钼;所述
除杂包括以下步骤:在搅拌的条件下,将氧化二硫化钼分散液进行冰浴,冰浴过程中在氧化二硫化钼分散液中加入50ml去离子水,搅拌30min后,停止冰浴;继续在氧化二硫化钼分散液中加入100ml去离子水,同时控制分散液温度在60℃以下;加入8ml质量浓度为30%的双氧水后,再加入250ml质量浓度为0.1%的盐酸;。
[0099]
将n,n
‑
二甲基乙酰胺和聚乙二醇400混合,按照转速为450r/min搅拌时间2h,得到混合溶液;将所述混合溶液和氧化二硫化钼依次进行超声和搅拌,得到分散液,其中超声的功率为500w,时间为4h,搅拌的转速为400r/min,时间为2h;将所述分散液和聚砜混合,按照转速为150r/min搅拌2h,得到铸膜液;铸膜液中聚乙二醇400的质量百分含量为1%,氧化二硫化钼的质量百分含量为0.03%,聚砜的质量百分含量为18%,n,n
‑
二甲基乙酰胺的质量百分含量为80.97%;
[0100]
将所述铸膜液在温度为50℃,真空度为0.8mpa的条件下静置4h时间后利用凹槽深度为100μm的刮刀在温度为25℃,相对湿度为50%的环境中将铸膜液涂覆在厚度为97μm密度为0.77g/m3的无纺布表面按照1.5m/min速度进行刮膜后在温度为80℃的空气中蒸发0.5min,然后将产品放入温度为25℃的自来水中固化0.5h后进行浸泡处理,所述浸泡处理按照以下步骤进行,将固化后产物放入温度为25℃的纯水中浸泡48h,然后放到质量浓度为30%温度为25℃的甘油水溶液中浸泡24h,最后将薄膜取出晾干,得到初级复合纳滤膜;
[0101]
配制水相溶液,其中樟脑磺酸的质量浓度为1.5%,三乙胺的质量浓度为1.5%,哌嗪的质量浓度为1.6%,十二烷基磺酸钠的质量浓度为0.6%;配置油相溶液,其中均苯三甲酰氯的质量和正己烷的体积比为0.35g:100ml;
[0102]
将所述初级复合纳滤膜在去离子水中浸泡2h后利用去离子水冲洗,将冲洗后的初级复合纳滤膜晾干;将晾干的初级复合纳滤膜在水相溶液中浸泡30s后取出,利用纸巾吸附纳滤膜表面多余的水相溶液后在油相溶液中浸泡20s,取出后利用纸巾吸附纳滤膜表面多余的油相溶液,60℃原位界面聚合反应2min,得到复合纳滤膜。
[0103]
实施例2~4
[0104]
按照实施例1的方法制备复合纳滤膜,不同之处在于,铸膜液的原料配比按照表1进行添加;
[0105]
表1实施例1~4和对比例1中铸膜液的原料配比
[0106][0107]
对比例1
[0108]
按照实施例1的方法制备复合纳滤膜,不同之处在于,铸膜液中不添加氧化二硫化钼,铸膜液中聚乙二醇400的质量百分含量为1%,聚砜的质量百分含量为18%,n,n
‑
二甲基乙酰胺的质量百分含量为81%。
[0109]
对比例2
[0110]
按照实施例1的方法制备复合纳滤膜,不同之处在于,不在基膜层表面进行界面聚合反应,即得到的复合纳滤膜不含有聚酰胺分离层。
[0111]
本发明根据gb/t 34242
‑
2017检测实施例1~4和对比例1、2制备的复合纳滤膜的纯水通量、孔径、接触角以及在0.4mpa压力下对硫酸钠、氯化钠和孟加拉玫瑰红的脱除率,其结果列于表2中。本发明在测试脱除率之前将实施例1~4和对比例1、2制备得到的复合纳滤膜在0.25mpa的压力预压1h。
[0112]
表2实施例1~4和对比例1、2制备得到的复合纳滤膜的性能
[0113][0114]
由表2的结果可知本发明提供的复合纳滤膜的纯水通量为13.4~27.7lm
‑2h
‑1bar
‑1,对硫酸钠的脱除率为84~95.6%,对孟加拉玫瑰红的脱除率为66.2~99.8%,具有良好的纯水通量和染料脱除率,本发明提供的复合纳滤膜具有良好的分离性能。
[0115]
实施例1和对比例1制备得到的复合纳滤膜的结构示意图,如图1所示。其中上面一条路线显示的是对比例1制备得到的基膜和复合纳滤膜的结构示意图;下面一条路线显示的是实施例1制备得到的基膜和复合纳滤膜的结构示意图。
[0116]
检测实施例2和对比例1制备得到的复合纳滤膜的膜孔径分布,得到膜孔径分布图,如图2所示。由图2可知实施例2制备得到的复合纳滤膜的孔径分布更窄,主要集中于0.1~0.5nm之间,因此表现出更好的截留性能。
[0117]
对实施例2和对比例1制备得到的复合纳滤膜的平面和断面进行扫描电镜检测,得到sem图如图3所示。从图3可以看出,与对照例1相比,实施例2制备得到的复合纳滤膜表面和断面均发生了较大的变化,本发明制备得到的复合纳滤膜表面出现较多的凸起,这是由于基膜表面存在较多的亲水性氧化二硫化钼片层结构。
[0118]
以工业稀土实际废水为待待处理废水,按照如下方法检测实施例2和对比例1制备得到的复合纳滤膜在分离实际废水中的长期运行情况:在0.4mpa压力下连续过滤稀土废水溶液,每隔1h记录一次通量;其结果列于表3中。
[0119]
表3实施例2和对比例1制备得到的复合纳滤膜过滤稀土废水的流通量
[0120][0121][0122]
根据表3的数据绘制实施例2和对比例1制备得到的复合纳滤膜长期运行通量对比曲线图,如图4所示。从图4可知,本发明提供的复合纳滤膜在稀土冶炼废水过滤长期运行过程中,其通量均高于对比例1的复合纳滤膜,本发明提供的复合纳滤膜具有较高的抗污染
性。
[0123]
尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例,而不是全部实施例,人们还可以根据本实施例在不经创造性前提下获得其他实施例,这些实施例都属于本发明保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1