一种羟丙基葡聚糖微球及其制备方法和用途与流程
1.本发明涉及分离纯化介质技术领域,特别是涉及一种羟丙基葡聚糖微球及其制备方法和用途。
背景技术:
2.近年来,随着中药技术的发展,开始采用层析技术来分离提取中药的有效成分。层析法是利用不同物质理化性质的差异而建立起来的技术。各组分不断地在两相中进行再分配。分部收集流出液,可得到样品中所含的各单一组分,从而达到将各组分分离的目的。相较于传统有效成分的提取,具有纯度高、污染小,耗时少等特点。
3.中药的化学成分极其复杂。传统中药多是煎熬后服用,有效成分多较为亲水,包括生物碱、黄酮、蒽醌、皂甙、有机酸、多糖、肽和蛋白质。灵活及综合性地利用层析方法。更容易分离到单一活性成分。
4.因此,开发出一种被指工艺简单易行,成本低,粒径可控的羟丙基葡聚糖微球及其制备方法具有重要意义。
技术实现要素:
5.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种羟丙基葡聚糖微球及其制备方法和用途,用于解决现有技术中的问题。
6.为实现上述目的及其他相关目的,本发明是通过包括以下技术方案获得的。
7.本发明提供一种羟丙基葡聚糖微球的制备方法,葡聚糖经交联剂活化形成葡聚糖微球,然后经羟丙基化处理后得到羟丙基葡聚糖微球。
8.优选地,活化反应在nabh4存在及碱性条件下进行。
9.优选地,采用葡聚糖水溶液与交联剂进行活化反应形成葡聚糖微球。
10.优选地,活化反应在搅拌条件下进行。
11.优选地,活化反应时,将交联剂滴加加入所述混合液中。优选地,交联剂的加入速度为0.1~2ml/min。如将交联剂采用蠕动泵泵入所述混合液,这种加入速度控制了反应过程中的放热,使得微球交联过程不易产生高温且稳定可行。
12.优选地,所述交联剂与葡聚糖水溶液的质量比为(1~8):(15~19)。
13.更优选地,交联剂的加入时间为3~6h。
14.优选地,葡聚糖水溶液中葡聚糖与水的质量比为(5~9):10。
15.优选地,在活化反应中,nabh4的加入量为葡聚糖含量为0.01~10wt%。
16.在活化反应中,碱性条件可以由氢氧化钠提供,碱性条件是指ph为8~14。
17.优选地,活化反应的反应温度为20~50℃。
18.优选地,所述交联剂为选自环氧氯丙烷、环氧溴丙烷或乙二醇二缩水甘油醚中的一种或多种。
19.优选地,对活化反应的产物进行后处理,以获得葡聚糖微球,所述葡聚糖微球的粒
径为30~120μm。
20.更优选地,所述后处理包括过滤、浓缩和干燥。更优选地,干燥温度为20~100℃。
21.更优选地,所述后处理还包括筛分。
22.优选地,羟丙基化处理时,将葡聚糖微球分散在含有甲苯、环氧烷、nabh4的碱性溶液中形成悬浮液,然后与配体进行羟丙基化反应。
23.更优选地,在进行羟丙基化反应时,碱性溶液的ph为8~14。更优选地,采用氢氧化钠调节ph。
24.更优选地,葡聚糖微球与甲苯的质量比为(1~2):10。
25.更优选地,环氧烷与甲苯的质量比为(1~2):2。
26.更优选地,nabh4的加入量为葡聚糖微球质量的0.01wt%~10wt%。
27.更优选地,所述配体与所述甲苯的质量比为(1~4):20。
28.优选地,羟丙基化处理的温度为20~50℃。
29.优选地,所述配体与所述葡聚糖微球的质量比为(1~10):20。
30.优选地,所述环氧烷为选自环氧乙烷、环氧丙烷和环氧丁烷中的一种或多种。
31.优选地,所述配体为选自苯扎溴铵、苄基三甲基氯化铵、苯扎氯氨和苄索氯氨中的一种或多种。
32.优选地,羟丙基处理还包括后处理步骤以获得纯净的羟基化葡聚糖微球。更优选地,所述后处理步骤包括过滤、浓缩和干燥。
33.本技术还公开了采用如上述所述的制备方法获得的羟丙基葡聚糖微球。
34.本技术还公开了如上述所述的羟丙基葡聚糖微球用于中药成分的分离纯化的用途。
35.本技术中,羟丙基葡聚糖微球是葡聚糖微球经过新型配体悬浮聚合后经功能化反应得到的产物,此时,葡聚糖微球中的葡聚糖部分将与羟丙基结合下形成醚键形式,与葡聚糖微球相比虽然羟基总数没变,但是碳原子数所占比例却增加了,使该介质同时具备亲水性和亲脂性,适合于中药有效成分的分离纯化。分离原理结合了凝胶过滤、反相层析的特点。在凝胶过滤的作用下,大分子化合物的保留力弱,先被洗脱下来,分子最小的最后出柱;在反相洗脱溶剂中,起反相分配作用,极性大的物质的保留弱先被洗脱,极性小的化合物后出柱。
36.本技术中,还对葡聚糖微球进行了改性,使葡聚糖微球接上具有带有生物活性的配基,即可用于不同生化物质的分离纯化,而且也解决了现有葡聚糖微球改性技术存在的制备工艺复杂、粒径不易控制、成本高等问题。
37.本技术上述技术方案具有以下有益效果:
38.(1)采用经过干燥筛分后的葡聚糖微球,使得羟丙基葡聚糖微球粒径更加均一可控,提高了产品的分辨率,产品具备亲水性和亲脂性,适合于中药有效成分的分离纯化,相比于传统市售羟丙基葡聚糖微球对茶叶中槲皮素的分离效果更佳。
39.(2)产品结合了凝胶过滤、反相层析的特点。在凝胶过滤的作用下,大分子化合物的保留力弱,先被洗脱下来,分子最小的最后出柱;在反相洗脱溶剂中,起反相分配作用,极性大的物质的保留弱先被洗脱,极性小的化合物后出柱。
40.(3)制备方法简单可行,无高温高转速工艺,有利于控制产品的稳定性,可放大生
产,能满足大规模生产需求。
附图说明
41.图1显示为采用实施例1中制备的羟丙基葡聚糖微球针对槲皮素粗品色谱图。
42.图2显示为采用实施例1中制备的羟丙基葡聚糖微球针对槲皮素纯度检定色谱图。
43.图3显示为采用市售羟丙基葡聚糖微球针对槲皮素粗品色谱图。
44.图4显示为采用市售羟丙基葡聚糖微球针对槲皮素纯度检定色谱图。
45.图5显示为实施例1中制备的羟丙基葡聚糖微球的粒径分析结果图。
46.图6显示为市售羟丙基葡聚糖微球的粒径分析效果图。
具体实施方式
47.以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。
48.在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。下列实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。
49.当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
50.本技术申请人基于要找到一种适用于中药成分或中药有效成分的分离纯化的微球,提供一种羟丙基葡聚糖微球。经制备和实验发现,其不仅制备方法简单,而且形成的羟丙基葡聚糖微球非常适合用于中药成分或中药有效成分的分离纯化。
51.在一个具体的羟丙基葡聚糖微球的制备方法中,采用葡聚糖经交联剂活化形成葡聚糖微球,然后经羟丙基化处理后得到羟丙基葡聚糖微球。
52.在本技术下述具体实施例中,所采用的一个更具体的制备方法为:
53.(1)在葡聚糖固体中加入一定量纯净水混合,加热,制得葡聚糖水溶液;
54.(2)葡聚糖水溶液中加入nabh4,再加入naoh溶液,并以100
‑
200rpm转速机械搅拌混合均匀;
55.(3)在搅拌条件下,按一定流速滴加交联剂,在一定温度下反应一定时间,滴加时间3
‑
6h。滴加完毕后再反应一定时间;
56.(4)将步骤(3)所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,葡聚糖微球的粒径为30~120μm;
57.(5)将干燥后的葡聚糖微球加入甲苯混合均匀,控温剧烈搅拌;
58.(6)将环氧烷缓慢加入步骤(5)溶液中,控温剧烈搅拌;
59.(7)向步骤(6)溶液中缓慢加入nabh4,再加入naoh溶液,控温剧烈搅拌;
60.(8)将步骤(7)溶液中缓慢加入配体,过夜反应,控温剧烈搅拌;
61.(9)将步骤(8)快速降温,滤布过滤、浓缩后干燥,制得羟丙基化葡聚糖微球。
62.为了说明本技术上述技术方案及其带来的技术效果,具体通过如下实施例加以说明和阐释。
63.实施例1
64.本实施例中羟丙基葡聚糖微球的制备方法:
65.称取500g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入5g的nabh4,加入50wt%naoh溶液50g,200rpm转速机械搅拌1h;称取100g的环氧氯丙烷用泵控制流速为0.5ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得500g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球500g加入2500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入1250g环氧丙烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入5g的nabh4,称取250g的50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,控温剧烈搅拌1h;称取125g苄索氯氨缓慢加入到溶液中,50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
66.采用本实施例中获得的羟丙基葡聚糖微球对槲皮素粗品进行分析的色谱图如图1所示,其中4号峰为槲皮素物质。
67.采用实施例1中制备的羟丙基葡聚糖微球针对槲皮素纯度检定色谱图如图2所示。
68.本实施例1中制备的羟丙基葡聚糖微球的粒径分析结果图如图5所示。
69.实施例2
70.本实施例中羟丙基葡聚糖微球的制备方法:
71.称取500g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入5g的nabh4,加入50%naoh溶液50g,200rpm转速机械搅拌1h;称取100g的环氧氯丙烷用泵控制流速为0.5ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得500g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球500g加入2500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入1250g环氧丙烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入5g的nabh4,称取250g的50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,,控温剧烈搅拌1h;称取112.5g苄基三甲基氯化铵缓慢加入到溶液中,50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
72.实施例3
73.本实施例中羟丙基葡聚糖微球的制备方法:
74.称取500g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入5g的nabh4,加入50%naoh溶液50g,200rpm转速机械搅拌1h;称取100g的环氧氯丙烷用泵控制流速为0.5ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得500g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球500g加入2500g甲苯混合均匀,
控温至50℃并500rpm剧烈搅拌,然后缓慢加入1250g环氧丙烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入5g的nabh4,称取250g的50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,,控温剧烈搅拌1h;称取137.5g苄基三甲基氯化铵缓慢加入到溶液中,50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
75.实施例4
76.本实施例中羟丙基葡聚糖微球的制备方法:
77.称取600g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入6.25g的nabh4,加入50%naoh溶液60g,200rpm转速机械搅拌1h;称取200g的环氧溴丙烷用泵控制流速为1ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得600g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球600g加入2500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入1500g环氧乙烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入6g的nabh4,称取300g的50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,控温剧烈搅拌1h;称取125g苯扎溴铵缓慢加入到溶液中,50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
78.实施例5
79.本实施例中羟丙基葡聚糖微球的制备方法:
80.称取600g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入6.25g的nabh4,加入50%naoh溶液60g,200rpm转速机械搅拌1h;称取200g的环氧溴丙烷用泵控制流速为1ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得600g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球600g加入2500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入1500g环氧乙烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入6g的nabh4,称取300g的50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,控温剧烈搅拌1h;称取112.5g苯扎溴铵缓慢加入到溶液中,50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
81.实施例6
82.本实施例中羟丙基葡聚糖微球的制备方法:
83.称取600g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入6.25g的nabh4,加入50%naoh溶液60g,200rpm转速机械搅拌1h;称取200g的环氧溴丙烷用泵控制流速为1ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得600g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球600g加入2500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入1500g环氧乙烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入6g的nabh4,称取300g的50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,控温剧烈搅拌1h;称取137.5g苯扎溴铵缓慢加入到溶液中,
50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
84.实施例7
85.本实施例中羟丙基葡聚糖微球的制备方法:
86.称取900g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入9.375g的nabh4,加入50%naoh溶液100g,200rpm转速机械搅拌1h;称取200g的乙二醇二缩水甘油醚用泵控制流速为1ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得900g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球900g加入4500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入2250g环氧丁烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入9g的nabh4,称取450g 50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,,控温剧烈搅拌1h;称取187.5g苯扎氯氨缓慢加入到溶液中,50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
87.本技术上述实施例中获得的羟丙基葡聚糖微球的粒径均在30~120μm的范围内,形成的产品具备亲水性和亲脂性,非常适合用于中药有效成分的分离纯化。
88.实施例8
89.本实施例中羟丙基葡聚糖微球的制备方法:
90.称取900g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入9.375g的nabh4,加入50%naoh溶液100g,200rpm转速机械搅拌1h;称取200g的乙二醇二缩水甘油醚用泵控制流速为1ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得900g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球900g加入4500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入2250g环氧丁烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入9g的nabh4,称取450g 50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,,控温剧烈搅拌1h;称取168.75g苯扎氯氨缓慢加入到溶液中,50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
91.本技术上述实施例中获得的羟丙基葡聚糖微球的粒径均在30~120μm的范围内,形成的产品具备亲水性和亲脂性,非常适合用于中药有效成分的分离纯化。
92.实施例9
93.本实施例中羟丙基葡聚糖微球的制备方法:
94.称取900g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入9.375g的nabh4,加入50%naoh溶液100g,200rpm转速机械搅拌1h;称取200g的乙二醇二缩水甘油醚用泵控制流速为1ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得900g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球900g加入4500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入2250g环氧丁烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入9g的nabh4,称取450g 50%naoh溶液用泵控制
流速为10ml/min,控温至50℃滴入糖溶液中,,控温剧烈搅拌1h;称取206.25g苯扎氯氨缓慢加入到溶液中,50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
95.本技术上述实施例中获得的羟丙基葡聚糖微球的粒径均在30~120μm的范围内,形成的产品具备亲水性和亲脂性,非常适合用于中药有效成分的分离纯化。
96.实施例10
97.本实施例中羟丙基葡聚糖微球的制备方法:
98.称取500g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入5g的nabh4,加入50%naoh溶液50g,200rpm转速机械搅拌1h;称取100g的环氧氯丙烷用泵控制流速为0.5ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得500g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球500g加入2500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入1250g环氧丙烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入5g的nabh4,称取250g的50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,,控温剧烈搅拌1h;称取112.5g苄索氯氨缓慢加入到溶液中,50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
99.实施例11
100.本实施例中羟丙基葡聚糖微球的制备方法:
101.称取500g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入5g的nabh4,加入50%naoh溶液50g,200rpm转速机械搅拌1h;称取100g的环氧氯丙烷用泵控制流速为0.5ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得500g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球500g加入2500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入1250g环氧丙烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入5g的nabh4,称取250g的50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,,控温剧烈搅拌1h;称取125g苄基三甲基氯化铵缓慢加入到溶液中,50℃下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
102.实施例12
103.本实施例中羟丙基葡聚糖微球的制备方法:
104.称取500g葡聚糖固体,加入到1000g的水中,加热到50℃,200rpm搅拌溶解2h,加入5g的nabh4,加入50%naoh溶液50g,200rpm转速机械搅拌1h;称取100g的环氧氯丙烷用泵控制流速为0.5ml/min,控温至50℃滴入糖溶液中,滴加完毕后,控温在50℃继续反应18h,停止加热,将所得溶液用滤布过滤、浓缩后干燥,制得葡聚糖微球,筛分后得500g葡聚糖微球,葡聚糖微球的粒径为30~120μm;称取干燥后的葡聚糖微球500g加入2500g甲苯混合均匀,控温至50℃并500rpm剧烈搅拌,然后缓慢加入1250g环氧丙烷,控温至50℃并500rpm剧烈搅拌,向溶液中缓慢加入5g的nabh4,称取250g的50%naoh溶液用泵控制流速为10ml/min,控温至50℃滴入糖溶液中,,控温剧烈搅拌1h;称取137.5g苄索氯氨缓慢加入到溶液中,50℃
下过夜反应,将所得溶液快速降温,滤布过滤、浓缩后80℃干燥12h,制得羟丙基化葡聚糖微球。
105.将上述实施例1~3葡聚糖微球用于茶叶中槲皮素的分离纯化。
106.1)分离纯化柱的制备:
107.制备:分别称取本实施例1中羟丙基葡聚糖微球和市售羟丙基葡聚糖微球100g填料,加入400ml的90%乙醇溶胀24h,得到约400ml的胶层和50ml上清层。用300ml容胀后填料装柱,得到1
×
15cm的柱体。装好柱后,用90%乙醇以2ml/min的流速平衡24h,平衡后上样。
108.再生:一次分离结束后,用80%的丙酮冲洗柱子,冲洗干净后用约两倍柱床体积的90%的乙醇平衡柱子,即可再次上样使用。
109.2)槲皮素的提取
110.称取50g茶样,置于2000ml烧杯中,加1500ml沸水,100℃水浴浸提40min,过滤;向滤渣中再加入1000ml沸水,100℃水浴浸提40min,过滤。滤液合并后,抽滤、浓缩后得浓缩液100ml。将浓缩液转移到分液漏斗中,分别用150ml、100ml、100ml氯仿萃取3次,脱除其中的部分杂质,然后水层继续用乙酸乙酯分别150ml、100ml、100ml萃取3次,合并酯层,经水浴50℃减压浓缩、真空干燥,得槲皮素粗品。
111.3)槲皮素样品的分离,纯化,检测;
112.称取1g 2)得到的槲皮素粗品,用10ml 9 0%乙醇溶解,0.45μm有机膜过滤后上1、中制备活化好的羟丙基葡聚糖柱分离。用90%乙醇以1ml/min的流速洗脱,每10ml收集一管。按照以下槲皮素纯度检定的hplc方法检测,以市售槲皮素标准品定性和外标法定量,合并含有相同槲皮素组分的试管,减压浓缩、真空干燥,4℃冰箱冷藏。
113.槲皮素纯度检定的hplc方法:色谱柱:月旭lp
‑
c18 4.6
×
250mm,5μm,流动相:甲醇/0.4%磷酸=50/50,检测波长:360nm,流速:1ml/min,柱温:30摄氏度;进样量:20μl,
114.市售槲皮素标准品:精密称取市售槲皮素标准品适量,用80%甲醇水溶解制成浓度是20μg/ml的槲皮素标准品溶液。
115.4)槲皮素粗品柱层析精制
116.将3)得到的槲皮素粗品,用10ml甲醇溶解后,上1)中制备纯化好的羟丙基葡聚糖柱上分离纯化,95%乙醇洗脱,洗脱速度为(1ml/min),每10ml收集一管,经液相检测,合并槲皮素组分,经减压浓缩、冷冻干燥后,得槲皮素纯品。按3)中的槲皮素纯度检定的hplc方法检测样品纯度,其纯度分别为98.2%、93.8%均满足市场需求,但是实施例1中的产品提取纯度更高。
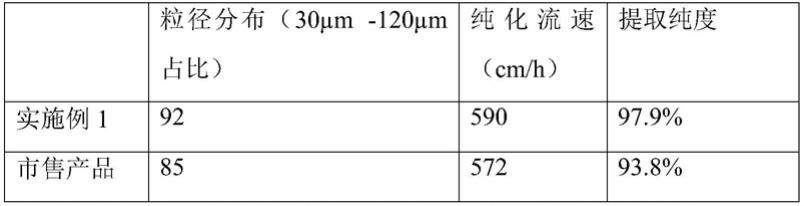
117.表1为实施例1和市售羟基葡聚糖微球的相关指标检测结果
[0118][0119]
相比市售产品实施例1中的产品粒径更加均匀,能适当提高纯化流速。
[0120]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1