一种过渡金属硫磷化物及其制备方法和应用、光催化分解水制氢用催化剂组合物
1.本发明属于催化剂助剂技术领域,特别涉及一种过渡金属硫磷化物及其制备方法和应用、光催化分解水制氢用催化剂组合物。
背景技术:
2.光催化分解水制氢是实现太阳能向化学能转换的绿色途径之一,对实现“碳中和”产生积极影响,开发高效的产氢催化剂助剂是提高光催化产氢效率的有效途径。过渡金属磷化物和过渡金属硫化物在光催化领域得到广泛研究,例如过渡金属硫磷化物作为光催化分解水制氢的催化剂助剂具有优异的产氢活性。
3.zhang等人(chemsuschem,2019,12(12):2651-2659)在管式炉中通过原位硫磷化法制备了具有光催化活性的磷硫铁多孔纳米片feps3光催化剂,在氙灯照射下,其表现出305.6mol
·
h-1
·
g-1
的光催化产氢速率;zhang等人(applied catalysis b.environmental,2020,273:118927)采用固态化学反应法和新型湿化学方法制备了两种不同结构的硫磷化铜cu3p|s和cus|p,cu3p|s的产氢速率达到2085μmol
·
g-1
·
h-1
,cus|p的产氢速率达到976μmol
·
g-1
·
h-1
;wang等人(advanced functional materials,2020,30(12):1908708)采用非金属掺杂提高层状nips3的产氢活性,其中c、n共掺杂nips3的催化性能与pt相当,其具有53.2mv的超低过电位,可提供10ma
·
cm-2
和0.7ma
·
cm-2
的高交换电流密度。
4.但目前的过渡金属硫磷化物制备过程复杂,工艺条件要求较高,具体的,如考虑将过渡金属磷化物和过渡金属硫化物同时沉积于半导体光催化剂表面,由于磷化物的制备方法一般采用高温热解或气相沉淀法,通常采用有机磷等有毒磷源以及惰性气体保护策略,致使过渡金属硫化物和过渡金属磷化物共同制备条件苛刻。具体来说,通常制备过渡金属硫磷化物通常采用有机磷等有毒磷源,需要在真空管式炉中,高温条件下采用惰性气体保护策略制备,耗时长产量低,成本高昂;而且很难同时引入磷源和硫源,特别是磷源和硫源的热分解温度不同,故很难同时得到过渡金属磷硫化物。
技术实现要素:
5.有鉴于此,本发明的目的在于提供一种过渡金属硫磷化物及其制备方法,本发明提供的制备方法可以实现原位同步制备含过渡金属硫化物和过渡金属磷化物的过渡金属硫磷化物,无需惰性气氛和高温条件,工艺简单温和;所得的过渡金属硫磷化物具有光催化活性优异的特点。
6.为了实现上述发明的目的,本发明提供以下技术方案:
7.本发明提供了一种过渡金属硫磷化物的制备方法,包括以下步骤:
8.将可溶性镍源、红磷、可溶性硫源和水混合,调节ph值,将所得的碱性原料液进行水热反应,得到所述过渡金属硫磷化物。
9.优选的,所述可溶性镍源中的镍元素与红磷中的磷元素的摩尔比为(2~8):(1.5~10);
10.所述可溶性镍源中的镍元素与可溶性硫源中的硫元素的摩尔比为(2~8):(0.25~6)。
11.优选的,所述可溶性镍源为氯化镍、硝酸镍或乙酸镍;
12.所述可溶性硫源为c2h5ns或ch4n2s。
13.优选的,所述碱性原料液的ph值为12~14。
14.优选的,所述水热反应的温度为140~180℃,时间为12~24h。
15.本发明还提供了上述技术方案所述制备方法得到的过渡金属硫磷化物,包括ni2p-nis颗粒。
16.优选的,所述ni2p-nis颗粒的化学组成包括ni2p和nis,所述ni2p和nis的摩尔比为15.5:(1~93)。
17.优选的,所述过渡金属硫磷化物的粒径为15~20nm。
18.本发明还提供了上述技术方案所述制备方法得到的过渡金属硫磷化物作为光催化分解水制氢催化剂助剂的应用。
19.本发明还提供了一种光催化分解水制氢用催化剂组合物,包括光催化剂和过渡金属硫磷化物;
20.所述光催化剂包括g-c3n4;
21.所述过渡金属硫磷化物为上述技术方案所述制备方法得到的过渡金属硫磷化物;
22.所述光催化分解水制氢用催化剂组合物中过渡金属硫磷化物的含量为3~25wt.%。
23.本发明提供了一种过渡金属硫磷化物的制备方法,包括以下步骤:将可溶性镍源、红磷、可溶性硫源和水混合,调节ph值,将所得的碱性原料液进行水热反应,得到所述过渡金属硫磷化物。本发明采用常规水热法同步制备得到过渡金属磷硫化物,磷源和硫源在液相环境中,能够在相对较小的空间中同时接触金属镍离子,同步生成过渡金属硫磷化物,整个制备过程无需惰性气氛、高温的苛刻条件,简单便捷,温和安全,成本低廉。本发明通过水热反应,一步生成的ni2p-nis结合更紧密,ni2p-nis之间交互生长而不是靠静电吸附,电子转移更有优势。
24.实施例测试结果表明,采用本发明提供的制备方法得到的过渡金属硫磷化物结构稳定,能很快转移光生电子,降低产氢过电位,提高表面产氢动力学。
附图说明
25.图1为本发明中光催化分解水制氢用催化剂组合物的光催化分解水制氢的机理图;
26.图2为实施例1和对比例1~2的产物的xrd图;
27.图3为实施例1所得过渡金属硫磷化物的tem图;
28.图4为应用例4和对比应用例1~4产物的xrd图;
29.图5为应用例4所得光催化分解水制氢用催化剂组合物的tem图;
30.图6为应用例1~6所得光催化分解水制氢用催化剂组合物和对比应用例1、对比应
用例3~4的产物的产氢速率图。
具体实施方式
31.本发明提供了一种过渡金属硫磷化物的制备方法,包括以下步骤:
32.将可溶性镍源、红磷、可溶性硫源和水混合,调节ph值,将所得的碱性原料液进行水热反应,得到所述过渡金属硫磷化物。
33.在本发明中,若无特殊说明,所述制备方法中的各组分均为本领域技术人员熟知的市售商品。
34.本发明将可溶性镍源、红磷、可溶性硫源和水混合,调节ph值,得到碱性原料液。
35.在本发明中,所述可溶性镍源优选为氯化镍、硝酸镍或乙酸镍,更优选为氯化镍。在本发明中,所述镍源对是否含结晶水没有限定。在本发明中,所述可溶性硫源优选为c2h5ns或ch4n2s,更优选为c2h5ns。
36.在本发明中,所述可溶性镍源中的镍元素与红磷中的磷元素的摩尔比优选为(2~8):(1.5~10),更优选为(2~7):(1.5~8),再优选为(2~5):(1.5~5)。在本发明中,所述可溶性镍源中的镍元素与可溶性硫源中的硫元素的摩尔比优选为(2~8):(0.25~6),更优选为(2~7):(0.4~4),再优选为(2~5):(0.5~3)。
37.在本发明中,所述水优选为去离子水。在本发明中,所述可溶性镍源中镍元素的摩尔量与水的体积的比优选为(2~8)mol:50l。
38.本发明对所述可溶性镍源、红磷、可溶性硫源和水的混合没有特殊限定,以能够将可溶性镍源、红磷、可溶性硫源和水混合均匀为准。
39.在本发明中,所述碱性原料液的ph值优选为12~14,更优选为12.5~13.5,最优选为13。
40.在本发明中,调节所述ph值的试剂优选为naoh水溶液。在本发明中,所述naoh水溶液的浓度优选为1~10mol/l,更优选为2~9mol/l,再优选为3~8mol/l。
41.得到碱性原料液后,本发明将所述碱性原料液进行水热反应,得到所述过渡金属硫磷化物。
42.在本发明中,所述水热反应的温度优选为140~180℃,更优选为140~170℃,再优选为140~160℃;时间优选为12~24h,更优选为12~20h,再优选为12~18h。在本发明中,所述水热反应的设备优选为配置有内衬的水热合成反应罐。在本发明中,所述水热合成反应罐中内衬的材质优选为聚四氟乙烯。
43.在本发明中,所述水热反应中发生的反应见式1~式4:
44.ni
2+
+2e
→
ni
ꢀꢀ
式1;
45.8p+12h2o
→
5ph3+3h3po4ꢀꢀ
式2;
46.3ni+2ph3→
ni2p+(ni-ph3)
ꢀꢀ
式3;
47.ni
2+
+s
2-→
nis
ꢀꢀ
式4。
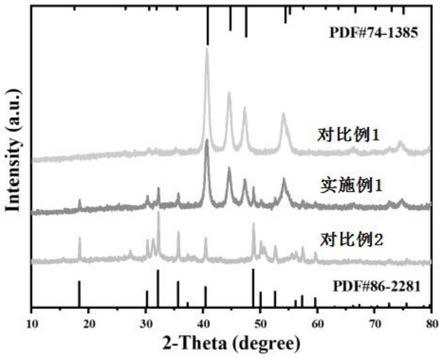
48.式1反应发生后,ni
2+
首先会吸附在rp的表面。随后,在水热的过程中,随着反应温度的升高,借助红磷自身的还原能力将ni
2+
还原。
49.所述水热反应后,本发明优选还包括:将水热反应产物依次进行清洗和干燥,得到所述过渡金属硫磷化物。
50.在本发明中,所述清洗优选为水洗离心。本发明对所述水洗离心没有特殊限定,采用本领域技术人员熟知的水洗离心即可。在本发明中,所述干燥的温度优选为60~70℃,更优选为60~68℃;时间优选为6~10h,更优选为7~10h。
51.在本发明中,所述过渡金属硫磷化物呈黑色。
52.本发明还提供了上述技术方案所述制备方法得到的过渡金属硫磷化物,包括ni2p-nis颗粒。
53.在本发明中,所述ni2p-nis颗粒的化学组成包括ni2p和nis,所述ni2p和nis的摩尔比优选为15.5:(1~93),更优选为15.5:(3~75),再优选为15.5:(5~50)。
54.在本发明中,所述过渡金属硫磷化物的粒径优选为15~20nm,更优选为16~19nm。
55.本发明还提供了上述技术方案所述制备方法得到的过渡金属硫磷化物作为光催化分解水制氢催化剂助剂的应用。
56.在本发明中,所述应用优选包括以下步骤:
57.将过渡金属硫磷化物和光催化剂混合,将所得的光催化分解水制氢用催化剂组合物进行光催化分解水制氢;
58.所述光催化剂包括g-c3n4;
59.所述光催化分解水制氢用催化剂组合物中过渡金属硫磷化物的含量为3~25wt.%。
60.本发明优选将过渡金属硫磷化物和光催化剂混合,将所得的光催化分解水制氢用催化剂组合物进行光催化分解水制氢。
61.在本发明中,所述光催化剂优选包括g-c3n4。在本发明中,所述g-c3n4优选为g-c3n4纳米片。
62.在本发明中,所述g-c3n4纳米片的制备方法优选包括以下步骤:
63.将碳氮化合物进行热聚合反应,得到g-c3n4颗粒;
64.将所述g-c3n4颗粒依次进行超声分散和干燥,得到所述g-c3n4纳米片。
65.本发明将碳氮化合物进行热聚合反应,得到g-c3n4颗粒。
66.在本发明中,所述碳氮化合物优选为尿素、氰胺、双氰胺、三聚氰胺和硫脲中的一种或多种。
67.在本发明中,所述热聚合反应优选为将碳氮化合物置于坩埚中,以锡纸包裹后,将包裹体系置于微波马弗炉中,在空气中升温至热聚合反应温度并保温。在本发明中,所述热聚合反应的温度优选为500~600℃,更优选为520~580℃;时间优选为2~4h,更优选为2.5~3.5h。在本发明中,升温至热聚合反应的升温速率优选为3~10℃/min,更优选为4~9℃/min。
68.热聚合反应后,本发明优选将热聚合反应产物进行研磨,得到g-c3n4颗粒。本发明对所述研磨没有特殊限定,以得到粉状的g-c3n4颗粒即可。
69.得到g-c3n4颗粒后,本发明将所述g-c3n4颗粒依次进行超声分散和干燥,得到所述g-c3n4纳米片。
70.本发明优选将所述g-c3n4颗粒分散于水中,进行超声分散。在本发明中,所述水优选为去离子水。本发明对所述超声分散的频率没有特殊限定,采用本领域技术人员熟知的超声频率即可。在本发明中,所述超声分散的时间优为10h。
71.在本发明中,所述干燥的温度优选为60~70℃,更优选为62~68℃;本发明对所述干燥的时间没有特殊限定,以能够将g-c3n4纳米片附着的水分去除为准。
72.在本发明中,所述过渡金属硫磷化物与上述技术方案所述过渡金属硫磷化物一致,在此不再赘述。
73.在本发明中,所述过渡金属硫磷化物和光催化剂的混合优选包括以下步骤:将过渡金属硫磷化物的分散悬浮液和光催化剂的分散悬浮液混合,搅拌蒸发去除过渡金属硫磷化物的分散悬浮液和光催化剂的分散悬浮液中的分散溶剂,得到光催化分解水制氢用催化剂组合物。
74.本发明优选将过渡金属硫磷化物和分散溶剂混合,得到所述过渡金属硫磷化物的分散悬浮液。在本发明中,所述过渡金属硫磷化物的分散悬浮液中的分散溶剂优选为乙醇。在本发明中,所述过渡金属硫磷化物的质量和分散溶剂的体积的比优选为(3~25)g:10l。在本发明中,所述过渡金属硫磷化物和分散溶剂的混合优选为超声。
75.本发明优选将光催化剂和分散溶剂混合,得到所述光催化剂的分散悬浮液。在本发明中,所述光催化剂的分散悬浮液中的分散溶剂优选为乙醇。在本发明中,所述光催化剂的质量和分散溶剂的体积的比优选为100g:10l。在本发明中,所述光催化剂和分散溶剂的混合优选为超声。
76.在本发明中,所述搅拌的速率优选为400~500rpm,更优选为440~460rpm。
77.在本发明中,所述光催化分解水制氢用催化剂组合物呈灰色。
78.本发明对所述光催化分解水制氢没有特殊限定,采用本领域技术人员熟知的光催化分解水制氢即可。
79.在本发明中,所述光催化分解水制氢用催化剂组合物的光催化分解水制氢的机理见图1。结合图1分析,在光照射下,光催化剂(g-c3n4)吸收光能后激发产生光生电子和空穴,电子跃迁至光催化剂(g-c3n4)的导带(cb)上,而空穴留在价带(vb)上;由于肖特基势垒的存在,电子能够“单向”不可逆地转移到过渡金属硫磷化物(ni2p-nis)上,从而快速将h
+
还原为h2,同时vb上的空穴氧化三乙醇胺(teoa),为体系持续提供电子,实现光催化分解水制氢。
80.本发明还提供了一种光催化分解水制氢用催化剂组合物,包括光催化剂和过渡金属硫磷化物;
81.所述光催化剂包括g-c3n4;
82.所述过渡金属硫磷化物为上述技术方案所述制备方法得到的过渡金属硫磷化物;
83.所述光催化分解水制氢用催化剂组合物中过渡金属硫磷化物的含量为3~25wt.%。
84.在本发明中,所述光催化分解水制氢用催化剂组合物中的物质组成与上述技术方案所述应用中的光催化分解水制氢用催化剂组合物一致,在此不再赘述。
85.为了进一步说明本发明,下面结合实施例对本发明提供的一种过渡金属硫磷化物及其制备方法和应用、光催化分解水制氢用催化剂组合物进行详细地描述,但不能将它们理解为对本发明保护范围的限定。显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
86.实施例1
87.将2mmol的nicl2·
6h2o、1.5mmol的红磷、0.5mmol的c2h5ns和50ml去离子水混合,利用10mol/l的naoh溶液调节ph值至13,磁力搅拌30min混合均匀,然后转移并密封在具有聚四氟乙烯内衬的水热合成反应罐中,于140℃下进行水热反应12h,将所得的水热反应产物冷却,水洗离心3次后于60℃干燥10h,得到黑色的过渡金属硫磷化物。
88.对比例1
89.将2mmol的nicl2·
6h2o、1.5的mmol红磷和50ml去离子水混合搅拌30min,将所得溶液体系转移并密封在具有聚四氟乙烯内衬的反应釜中,于140℃下进行水热反应12h,离心干燥,得到ni2p纳米颗粒。
90.对比例2
91.将1mmol的ni(ch3coo)2·
4h2o和50ml乙醇混合搅拌30min并超声处理20min,将所得的溶液体系和1mmol硫脲混合搅拌30min,于200℃下进行水热反应12h,离心干燥,得到nis纳米颗粒。
92.对实施例1和对比例1~2的产物进行x射线衍射测试,所得xrd图见图2。由图2可见,实施例1制备得到的过渡金属硫磷化物(ni2p-nis)的xrd图谱对应于ni2p和nis的两个标准卡片(jcpds pdf#74-1385,jcpds pdf#86-2281),其中40.7
°
、44.6
°
、47.4
°
和54.2
°
的衍射峰属于ni2p(jcpds pdf#74-1385)的(111)、(201)、(210)和(300)晶面,18.4
°
、32.2
°
、35.7
°
、40.5
°
、48.8
°
和52.6
°
分别对应于nis(jcpds pdf#86-2281)的(110)、(300)、(021)、(211)、(131)和(401)晶面。
93.对实施例1所得过渡金属硫磷化物进行透射电镜测试,所得tem图见图3,图3中,(a)为过渡金属硫磷化物的tem图,(b)为过渡金属硫磷化物的hrtem图。由图3可以清晰地看到间距为0.191nm和0.222nm的晶格条纹,分别对应于ni2p的(210)面和nis的(211)面,这与xrd结果一致。
94.应用例1
95.将100mg的g-c3n4纳米片和10ml乙醇超声混合2h,得到光催化剂的分散悬浮液;将3mg实施例1制备的过渡金属硫磷化物ni2p-nis颗粒与10ml乙醇超声混合2h,得到过渡金属硫磷化物的分散悬浮液;将光催化剂的分散悬浮液和过渡金属硫磷化物的分散悬浮液混合,连续搅拌蒸发去除乙醇,得到灰色粉末状的光催化分解水制氢用催化剂组合物(记为3%ni2p-nis/g-c3n4)。
96.应用例2
97.实施例1制备的过渡金属硫磷化物ni2p-nis颗粒的质量为5mg,其余技术手段与应用例1一致,得到应用例2(记为5%ni2p-nis/g-c3n4)。
98.应用例3
99.实施例1制备的过渡金属硫磷化物ni2p-nis颗粒的质量为10mg,其余技术手段与应用例1一致,得到应用例3(记为10%ni2p-nis/g-c3n4)。
100.应用例4
101.实施例1制备的过渡金属硫磷化物ni2p-nis颗粒的质量为15mg,其余技术手段与应用例1一致,得到应用例4(记为15%ni2p-nis/g-c3n4)。
102.应用例5
103.实施例1制备的过渡金属硫磷化物ni2p-nis颗粒的质量为20mg,其余技术手段与
应用例1一致,得到应用例5(记为20%ni2p-nis/g-c3n4)。
104.应用例6
105.实施例1制备的过渡金属硫磷化物ni2p-nis颗粒的质量为25mg,其余技术手段与应用例1一致,得到应用例6(记为25%ni2p-nis/g-c3n4)。
106.对比应用例1
107.仅g-c3n4纳米片。
108.对比应用例2
109.仅过渡金属硫磷化物ni2p-nis颗粒。
110.对比应用例3
111.以15mg对比例1制备的ni2p纳米颗粒代替应用例1中3mg实施例1制备的过渡金属硫磷化物ni2p-nis颗粒,其余技术手段与应用例1一致,得到对比应用例3(记为15%ni2p/g-c3n4)。
112.对比应用例4
113.以15mg对比例2制备的nis纳米颗粒代替应用例1中3mg实施例1制备的过渡金属硫磷化物ni2p-nis颗粒,其余技术手段与应用例1一致,得到对比应用例4(记为15%nis/g-c3n4)。
114.对应用例4和对比应用例1~4产物进行x射线衍射测试,所得xrd图见图4。由图4可见,纯g-c3n4的xrd图谱,27.4
°
和13.0
°
分别对应g-c3n4(jcpds pdf#87-1526)的(100)和(002)晶面;另外,加载ni2p-nis后g-c3n4的主衍射峰保存完好,说明g-c3n4在溶剂蒸发过程中没有被破坏。
115.对应用例4所得光催化分解水制氢用催化剂组合物进行投射电镜测试,所得tem图见图5,图5中,(a)为应用例4所得光催化分解水制氢用催化剂组合物的tem图,(b)为应用例4所得光催化分解水制氢用催化剂组合物的hrtem图,(c)为图5中(b)的局部放大图。由图5可见,光催化分解水制氢用催化剂组合物中ni2p-nis纳米颗粒成功被加载到g-c3n4的表面,0.220nm和0.195nm的晶格条纹分别来自nis的(211)面和ni2p的(210)面,这也与xrd结果一致。
116.对应用例1~6所得光催化分解水制氢用催化剂组合物和对比应用例1、对比应用例3~4的产物进行催化制氢测试,测试方法为:在labsolar-iiiag型光催化分解水产氢装置中进行测定,光源为300w氙灯(cel-hxf300),照明面积为24cm2,产氢前通过光辐射计(pl-mw2000)测量光照强度;具体操作如下:首先配置20vol%的牺牲剂混合液,内含80ml去离子水和20ml三乙醇胺(teoa);然后将10mg待测样品分散于牺牲剂中,将悬浮液放入反应器中进行磁力搅拌;搭好装置后,抽真空除去装置内部和牺牲剂混合液中的空气;最后开始光照进行产氢实验,产氢速率每隔1h测定一次;所得产氢速率图见图6。
117.由图6可见,应用例4提供的光催化分解水制氢用催化剂组合物(15w%ni2p-nis/g-c3n4)的产氢率最高,为6892.7μmol
·
g-1
·
h-1
,比对比应用例1提供的g-c3n4纳米片(150μmol
·
g-1
·
h-1
)提高了46.1倍;而且应用例4提供的光催化分解水制氢用催化剂组合物(15w%ni2p-nis/g-c3n4)的产氢率比对比应用例3中15w%ni2p/g-c3n4和对比应用例4中15w%nis/g-c3n4的产氢率分别高出4.4倍和7.5倍,说明本发明提供的过渡金属硫磷化物(ni2p-nis颗粒)作为光催化析氢反应的催化剂助剂比单独使用ni2p或nis更好。
118.以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1