一种催化裂解气脱氧催化剂、其制备方法和裂解气脱氧的方法与流程
1.本发明涉及气体净化技术领域,尤其是涉及一种催化裂解气脱氧催化剂、其制备方法和裂解气脱氧的方法。
背景技术:
2.乙烯和丙烯是聚合材料的关键基础原料,含有微量氧杂质会严重影响聚合催化剂和合成材料性能,通常在聚合工艺前需要对乙烯和丙烯脱氧净化处理到微量氧含量小于1x10-6
。随着新技术发展,不断出现轻烃催化裂解工艺替代蒸汽裂解来增产乙烯和丙烯,为了后续乙烯和丙烯聚合使用必须对催化裂解气进行脱氧净化处理。
3.脱氧剂已广泛应用于各种煤制合成气、天然气、高纯气、精细化工及聚酯、电子等工业气中氧的脱除。按照脱氧机理,主要分为催化脱氧和化学吸附脱氧两大类,其中催化脱氢主要分为耗炭、耗h2、耗co、耗烃等脱氧机理。工业应用的耗氢脱氧催化剂主要以pt、pd为活性组分的贵金属负载催化剂,这类脱氧剂脱氧活性高,脱氧容量大,但催化剂价格昂贵,装置投资成本高且易发生加氢副反应。化学吸附类的脱氧催化剂主要为铜系和锰系,利用低价态的金属对氧气进行化学吸附反应,这类脱氧剂常用到的原料气中不含氢气或含有微量氢气,不会发生加氢副反应,但是比较适合500x10-6
以下的低氧含量的原料气,脱氧容量小,再生频繁,造成工业运行成本较高。催化裂解气含有高含量的氢气、烯烃、0.1%以下氧气,因此,开发催化剂活性高,不损失烯烃含量且寿命长的脱氧催化剂是必须的。
4.单一活性组份cuo脱氧剂,耐热温度很低,200℃上就会发生烧结,形成较大晶粒而失去活性。使用过程中还原再生为放热反应,控制不佳导致床层极易超过200℃,因而还原再生时,容易发生烧结而失去脱氧活性;单一活性组份mno2脱氧剂,还原再生时的温度较高,通常超过300℃,消耗热能大,不利于大规模工业应用。
5.我国烯烃下游高分子材料飞速发展,对乙烯和丙烯的需求逐年激增。近年来轻烃催化裂化多产烯烃的工艺开始逐渐替代蒸汽裂解制乙烯工业,正在推广应用,而目前与工艺匹配的催化裂解气脱氧催化剂还没有深度研究开发。
6.专利cn101745391公开了一种以pd为主活性组分,ag、au、co、cr为助活性组分的脱氧剂,用于催化裂化干气脱除微量氧,但催化剂为昂贵的贵金属催化剂,同时会造成乙烯气体0.5%-1.7%的损失。专利cn103157471公开了一种以pd、pt及ag为主活性组分,用于烯烃气体中10000ppm以下氧脱除,并且避免了烯烃的损失,但是同样存在催化剂为昂贵的贵金属缺点,并且原料气中氢气含量仅为0.2%,不太适合高含量氢气的烯烃原料气。专利cn108620063公开了一种以mn主活性组分,加入稀土金属和粘结剂,用于炼厂干气脱除微量氧。该脱氧剂适用炼厂干气中100-200ppm氧气脱除,原料氧含量增高势必会导致脱氧剂再生频繁。专利cn101165030a公开了一种以mn-ag为双活性组分,用于乙烯脱氧净化,而处理方式为室温化学吸附机理,在处理高氧含量时,仍然会面临再生频繁问题。
技术实现要素:
7.有鉴于此,本发明要解决的技术问题在于提供一种催化裂解气脱氧催化剂,本发明制备的催化剂还原温度低,低温活性好,脱氧精度高以及寿命长,使用过程中确保烯烃不被加成损失。本发明适用于富含氢气的烃类原料净化脱氧,特别适用催化裂解气的氧气脱除。
8.本发明提供了一种催化裂解气脱氧催化剂,原料包括:20~60重量份的活性组分、10~30重量份的助剂和26~65重量份的载体;其中活性组分包括cuo和mn3o4;助剂为zno;载体为碱土金属氧化物改性的tio2/al2o3复合氧化物。
9.优选的,所述活性组分中cuo为2~10重量份,mn3o4为20~50重量份。
10.优选的,所述载体中碱土金属氧化物为10~25重量份,tio2为1~5重量份,al2o3为15~35重量份;所述碱土金属氧化物选自mgo、cao和bao中的一种或两种。
11.本发明提供了一种催化裂解气脱氧催化剂的制备方法,包括:
12.a)tio2前驱体和al2o3前驱体置于去离子水中,配制成tio2/al2o3悬浊液;
13.b)取cuo前驱体、mn3o4前驱体、zno前驱体以及碱土金属氧化物对应的前驱体与水混合并搅拌均匀,得到a溶液;
14.取碳酸钠、氢氧化钠,与水混合并搅拌均匀,得到b溶液;
15.c)在搅拌条件下,将制备的a溶液和b溶液滴加入到所述悬浊液中,发生共沉淀反应,陈化,沉淀物洗涤、干燥、焙烧、压片即得用于催化裂解气脱氧催化剂。
16.优选的,所述tio2前驱体为tio2粉或偏钛酸;所述al2o3前驱体为al2o3粉或者选自氢氧化铝干胶或拟薄水铝石;所述cuo前驱体为硝酸铜;所述mn3o4前驱体为乙酸锰或硝酸锰;所述zno前驱体为硝酸锌。
17.优选的,步骤b)中碳酸钠、氢氧化钠和水的质量比为(3.5~8.0):1:(25~40);
18.步骤c)所述共沉淀过程的ph值为7~8;共沉淀时间为1~4h。
19.优选的,所述洗涤具体为:洗涤至中性或滤液中检测不到na
+
为基准;所述的干燥温度为80~120℃、时间8~12h;所述的焙烧具体为:以3~8℃/min的升温速率升至450~550℃,焙烧2~6h。
20.本发明提供了一种裂解气脱氧的方法,包括:
21.上述技术方案任一项所述或上述技术方案任一项制备方法制备得到的催化裂解气脱氧催化剂在反应器中装填、干燥及还原后,含氧原料气与脱氧催化剂反应,即得。
22.优选的,所述装填为密相装填,催化剂颗粒大小2~8mm,装填高径比大于2;
23.所述含氧原料气为石脑油催化裂化后的裂解气;所述裂解气包括氢气、甲烷、乙烷、乙烯、乙炔、丙烷、丙烯、c4~c6的烷烃、c4~c6的烯烃、微量氧气、一氧化碳或二氧化碳中的一种或多种。
24.优选的,所述脱氧催化剂还原条件:氢气120℃干燥1~3h,还原气体为氢气,氢气体积空速为500~1000h-1
,还原温度为200~300℃,还原时间为2~5h;
25.所述反应条件:体积空速1000~5000h-1
,反应温度100~180℃,反应压力0.1~2.0mpa。
26.与现有技术相比,本发明提供了一种催化裂解气脱氧催化剂,原料包括:20~60重量份的活性组分、10~30重量份的助剂和26~65重量份的载体;其中活性组分包括cuo和
mn3o4;助剂为zno;载体为碱土金属氧化物改性的tio2/al2o3复合氧化物。本发明采用共沉淀制备工艺,存在活性组分含量和机械强度高等特点;在共沉淀过程中将碱土金属氧化物沉积在载体tio2/al2o3表面,覆盖了一部分酸性位从而实现载体改性,改性后使催化剂具有更好的低温反应性能。此外采用并流共沉淀的形式,将活性组分cuo、mn3o4主要沉淀在zno和碱土金属氧化物上,而非沉淀在载体tio2/al2o3表面,从而催化剂具有更高的分散度和热稳定性,以及活性中心在焙烧、还原和反应过程中具有更高抗烧结性能等优势;同时tio2/al2o3载体经过碱土金属改性修饰,使其具有合适的酸碱性,提高目标产物的选择性,降低烯烃损失。此外利用催化耗氢和化学吸附机理,实现应用条件范围广,使用周期长。
具体实施方式
27.本发明提供了一种催化裂解气脱氧催化剂、其制备方法和裂解气脱氧的方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都属于本发明保护的范围。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
28.本发明提供了一种催化裂解气脱氧催化剂,原料包括:20~60重量份的活性组分、10~30重量份的助剂和26~65重量份的载体;其中活性组分包括cuo和mn3o4;所述助剂为zno;所述载体为碱土金属氧化物改性的tio2/al2o3复合氧化物。
29.本发明所述重量份在总量为100时,等同于质量百分比。
30.本发明提供的催化裂解气脱氧催化剂,原料包括:20~60重量份的活性组分;其中,其中活性组分包括cuo和mn3o4;所述活性组分中cuo为2~10重量份,mn3o4为20~50重量份;优选的,所述活性组分中cuo为3~9重量份,mn3o4为25~45重量份;
31.本发明提供的催化裂解气脱氧催化剂,原料包括:10~30重量份的助剂;优选包括12~28重量份的助剂;更优选包括15~25重量份的助剂;所述助剂为zno。
32.本发明提供的催化裂解气脱氧催化剂,原料包括:26~65重量份的载体;所述载体为碱土金属氧化物改性的tio2/al2o3复合氧化物。
33.优选的,本发明所述载体中碱土金属氧化物为10~25重量份,tio2为1~5重量份,al2o3为15~35重量份;更优选的,所述载体中碱土金属氧化物为12~22重量份,tio2为1~4重量份,al2o3为18~32重量份。所述碱土金属氧化物选自mgo、cao和bao中的一种或两种。
34.本发明所述的各组分物质来源于其纯物质或前驱体,前驱体分别选自:
35.所述cuo、zno和碱土金属氧化物前驱体为其对应的硝酸盐;
36.所述mn3o4前驱体为乙酸锰和硝酸锰的任意一种;
37.所述al2o3可直接选用al2o3粉或者选自氢氧化铝干胶或拟薄水铝石作为前驱体;而所选用前驱体的比表面积大于300m2/g,孔容大于0.5cm3/g,干基≥68wt%;
38.所述tio2可直接选用tio2粉或者选自偏钛酸作为前驱体,其中偏钛酸的比表面积大于200m2/g,干基≥75wt%。
39.本发明提供了一种催化裂解气脱氧催化剂的制备方法,包括:
40.a)tio2前驱体和al2o3前驱体置于去离子水中,配制成tio2/al2o3悬浊液;
41.b)取cuo前驱体、mn3o4前驱体、zno前驱体以及碱土金属氧化物对应的前驱体与水
混合并搅拌均匀,得到a溶液;
42.取碳酸钠、氢氧化钠,与水混合并搅拌均匀,得到b溶液;
43.c)在搅拌条件下,将制备的a溶液和b溶液滴加入到所述悬浊液中,发生共沉淀反应,陈化,沉淀物洗涤、干燥、焙烧、压片即得用于催化裂解气脱氧催化剂。
44.本发明提供的一种催化裂解气脱氧催化剂的制备方法首先将tio2前驱体和al2o3前驱体置于去离子水中,配制成tio2/al2o3悬浊液。
45.优选具体为:依次按照催化剂中tio2、al2o3的含量,分别称取tio2粉或偏钛酸、al2o3粉或与其对应的前驱体,放入准备的去离子水中配制成悬浊液,搅拌并使其分散均一。本发明对于所述搅拌的具体方式不进行限定,本领域技术人员熟知的即可。本发明对于上述tio2、al2o3的含量和配比上述已经有了清楚的描述,在此不再赘述。
46.取cuo前驱体、mn3o4前驱体、zno前驱体以及碱土金属氧化物对应的前驱体与水混合并搅拌均匀,得到a溶液;本发明对于所述搅拌的具体方式不进行限定,本领域技术人员熟知的即可。
47.上述的各组分物质来源于其纯物质或前驱体,所述的前驱体分别选自:cuo、zno和碱土金属氧化物前驱体为其对应的硝酸盐;mn3o4前驱体为乙酸锰和硝酸锰的任意一种;al2o3可直接选用al2o3粉或者选自氢氧化铝干胶或拟薄水铝石作为前驱体;tio2可直接选用tio2粉或者选自偏钛酸作为前驱体。
48.进一步地,所述的al2o3前驱体的比表面积大于300m2/g,孔容大于0.5cm3/g,干基≥68wt%;
49.进一步地,所述的tio2前驱体的比表面积大于200m2/g,干基≥75wt%。
50.取碳酸钠、氢氧化钠,与水混合并搅拌均匀,得到b溶液;
51.本发明碳酸钠、氢氧化钠和水的质量比优选为(3.5~8.0):1:(25~40);更优选为(4~7.5):1:(28~38)。
52.本发明采用的是碳酸钠、氢氧化钠和水以上述比例配合获得的沉淀剂,这种混合沉淀剂可以使活性组分分散更均匀,且热稳定性高,在焙烧、还原和反应过程中避免氧化铜晶粒烧结而造成活性降低。
53.在搅拌条件下,将制备的a溶液和b溶液滴加入到所述悬浊液中,发生共沉淀反应,陈化。
54.本发明在搅拌条件下,将制备的a、b溶液缓慢滴加入到上述获得的悬浊液中,控制整个沉淀过程中ph=7~8,沉淀时间控制为1~3h。
55.沉淀过程结束后,在60~90℃下继续搅拌4~8h,进行陈化过程。
56.沉淀物洗涤、干燥、焙烧、压片即得用于催化裂解气脱氧催化剂。
57.进一步地,本发明所述洗涤优选具体为:洗涤至中性或滤液中检测不到na
+
为基准;
58.所述的干燥温度优选为80~120℃、时间8~12h;更优选为90~120℃、时间8~11h;
59.所述的焙烧优选具体为:以3~8℃/min的升温速率升至450~550℃,焙烧2~6h;更优选具体为:以4~8℃/min的升温速率升至460~540℃,焙烧3~6h。
60.本发明对于压片的具体操作不进行限定,本领域技术人员熟知的即可。
61.本发明催化剂采用共沉淀制备方法,具有活性位分布均匀,制备工艺简单的优势;同时,所述催化剂还原温度低,低温活性好,脱氧精度高以及寿命长,使用过程中确保烯烃不被加成损失。本发明适用于富含氢气的烃类原料净化脱氧,特别适用催化裂解气的氧气脱除。
62.本发明采用共沉淀制备工艺,存在活性组分含量和机械强度高等特点;在共沉淀过程中将碱土金属氧化物沉积在载体tio2/al2o3表面,覆盖了一部分酸性位从而实现载体改性,改性后使催化剂具有更好的低温反应性能。此外采用并流共沉淀的形式,将活性组分cuo、mn3o4主要沉淀在zno和碱土金属氧化物上,而非沉淀在载体tio2/al2o3表面,从而催化剂具有更高的分散度和热稳定性,以及活性中心在焙烧、还原和反应过程中具有更高抗烧结性能等优势;同时tio2/al2o3载体经过碱土金属改性修饰,使其具有合适的酸碱性,提高目标产物的选择性,降低烯烃损失。此外利用催化耗氢和化学吸附机理,实现应用条件范围广,使用周期长。
63.本发明提供了一种裂解气脱氧的方法,包括:
64.上述技术方案任一项所述或上述技术方案任一项制备方法制备得到的催化裂解气脱氧催化剂在反应器中装填、干燥及还原后,含氧原料气与脱氧催化剂反应,即得。
65.优选的,本发明所述催化剂在固定床反应器中装填、干燥及还原后,在一定反应条件下将含氧原料气从反应器上端经过脱氧催化剂,并通过在线微量检测仪检测尾气中氧气含量。
66.其中,所述装填为密相装填,催化剂颗粒大小2~8mm,装填高径比大于2;
67.所述含氧原料气为石脑油催化裂化后的裂解气;所述裂解气包括氢气、甲烷、乙烷、乙烯、乙炔、丙烷、丙烯、c4~c6的烷烃、c4~c6的烯烃、微量氧气、一氧化碳或二氧化碳中的一种或多种。
68.本发明所述脱氧催化剂还原条件:氢气120℃干燥1~3h,还原气体为氢气,氢气体积空速为500~1000h-1
,还原温度为200~300℃,还原时间为2~5h;
69.具体的,所述反应条件:体积空速1000~5000h-1
,反应温度100~180℃,反应压力0.1~2.0mpa。
70.本发明提供了一种催化裂解气脱氧催化剂,原料包括:20~60重量份的活性组分、10~30重量份的助剂和26~65重量份的载体;其中活性组分包括cuo和mn3o4;助剂为zno;载体为碱土金属氧化物改性的tio2/al2o3复合氧化物。本发明采用共沉淀制备工艺,存在活性组分含量和机械强度高等特点;在共沉淀过程中将碱土金属氧化物沉积在载体tio2/al2o3表面,覆盖了一部分酸性位从而实现载体改性,改性后使催化剂具有更好的低温反应性能。此外采用并流共沉淀的形式,将活性组分cuo、mn3o4主要沉淀在zno和碱土金属氧化物上,而非沉淀在载体tio2/al2o3表面,从而催化剂具有更高的分散度和热稳定性,以及活性中心在焙烧、还原和反应过程中具有更高抗烧结性能等优势;同时tio2/al2o3载体经过碱土金属改性修饰,使其具有合适的酸碱性,提高目标产物的选择性,降低烯烃损失。此外利用催化耗氢和化学吸附机理,实现应用条件范围广,使用周期长。
71.为了进一步说明本发明,以下结合实施例对本发明提供的一种催化裂解气脱氧催化剂、其制备方法和裂解气脱氧的方法进行详细描述。
72.实施例1
73.称取1.18g偏钛酸、25.00g氢氧化铝干胶,加水1l配制成氢氧化铝干胶悬浊液配制,在60℃下搅拌并使其分散均一。称取4.76g硝酸铜、162.29g乙酸锰、36.92g硝酸锌、74.19g硝酸镁,与3l水混合并搅拌均匀。称取640.02g碳酸钠、106.67g氢氧化钠,与3l水混合并搅拌均匀。在搅拌条件下,向氢氧化铝干胶悬浊液中同时加入上述硝酸盐溶液和碱溶液,控制整个过程为4小时,ph控制7-8。形成的沉淀经过经过过滤、水洗涤至中性或滤液中检测达不到na
+
、110℃干燥10h、540℃焙烧4h、压片成形等步骤得到脱氧催化剂c-1。
74.实施例2
75.称取3.53g偏钛酸、44.12g氢氧化铝干胶,加水1l配制成氢氧化铝干胶悬浊液配制,在60℃下搅拌并使其分散均一。称取4.76g硝酸铜、113.60g乙酸锰、44.30g硝酸锌、66.77g硝酸镁,与3l水混合并搅拌均匀。称取640.02g碳酸钠、106.67g氢氧化钠,与3l水混合并搅拌均匀。在搅拌条件下,向氢氧化铝干胶悬浊液中同时加入上述硝酸盐溶液和碱溶液,控制整个过程为4小时,ph控制7-8。形成的沉淀经过经过过滤、水洗涤至中性或滤液中检测达不到na
+
、110℃干燥10h、540℃焙烧4 h、压片成形等步骤得到脱氧催化剂c-2。
76.实施例3
77.称取2.35g偏钛酸、29.31g氢氧化铝干胶,加水1l配制成氢氧化铝干胶悬浊液配制,在70℃下搅拌并使其分散均一。称取7.14g硝酸铜、129.83g乙酸锰、55.38g硝酸锌、74.19g硝酸镁,与3l水混合并搅拌均匀。称取640.02g碳酸钠、106.67g氢氧化钠,与3l水混合并搅拌均匀。在搅拌条件下,向氢氧化铝干胶悬浊液中同时加入上述硝酸盐溶液和碱溶液,控制整个过程为4小时,ph控制7-8。形成的沉淀经过经过过滤、水洗涤至中性或滤液中检测达不到na
+
、120℃干燥8h、540℃焙烧4h、压片成形等步骤得到脱氧催化剂c-3。
78.实施例4
79.称取5.88g偏钛酸、44.12g氢氧化铝干胶,加水1l配制成氢氧化铝干胶悬浊液配制,在60℃下搅拌并使其分散均一。称取7.14g硝酸铜、81.14g乙酸锰、73.84g硝酸锌、63.06g硝酸镁,与3l水混合并搅拌均匀。称取640.02g碳酸钠、106.67g氢氧化钠,与3l水混合并搅拌均匀。在搅拌条件下,向氢氧化铝干胶悬浊液中同时加入上述硝酸盐溶液和碱溶液,控制整个过程为4小时,ph控制7-8。形成的沉淀经过经过过滤、水洗涤至中性或滤液中检测达不到na
+
、110℃干燥10h、500℃焙烧5h、压片成形等步骤得到脱氧催化剂c-4。
80.实施例5
81.称取3.53g偏钛酸、36.76g氢氧化铝干胶,加水1l配制成氢氧化铝干胶悬浊液配制,在70℃下搅拌并使其分散均一。称取11.91g硝酸铜、113.60g乙酸锰、44.30g硝酸锌、74.19g硝酸镁,与3l水混合并搅拌均匀。称取640.02g碳酸钠、106.67g氢氧化钠,与3l水混合并搅拌均匀。在搅拌条件下,向氢氧化铝干胶悬浊液中同时加入上述硝酸盐溶液和碱溶液,控制整个过程为4小时,ph控制7-8。形成的沉淀经过经过过滤、水洗涤至中性或滤液中检测达不到na
+
、110℃干燥10h、540℃焙烧4 h、压片成形等步骤得到脱氧催化剂c-5。
82.实施例6
83.称取2.35g偏钛酸、26.47g氢氧化铝干胶,加水1l配制成氢氧化铝干胶悬浊液配制,在60℃下搅拌并使其分散均一。称取11.91g硝酸铜、146.06g乙酸锰、73.84g硝酸锌、37.10g硝酸镁,与3l水混合并搅拌均匀。称取640.02g碳酸钠、106.67g氢氧化钠,与3l水混合并搅拌均匀。在搅拌条件下,向氢氧化铝干胶悬浊液中同时加入上述硝酸盐溶液和碱溶
液,控制整个过程为4小时,ph控制7-8。形成的沉淀经过经过过滤、水洗涤至中性或滤液中检测达不到na
+
、100℃干燥12h、540℃焙烧4 h、压片成形等步骤得到脱氧催化剂c-6。
84.实施例7
85.称取3.53g偏钛酸、36.76g氢氧化铝干胶,加水1l配制成氢氧化铝干胶悬浊液配制,在60℃下搅拌并使其分散均一。称取16.67g硝酸铜、129.83g乙酸锰、36.92g硝酸锌、55.64g硝酸镁,与3l水混合并搅拌均匀。称取640.02g碳酸钠、106.67g氢氧化钠,与3l水混合并搅拌均匀。在搅拌条件下,向氢氧化铝干胶悬浊液中同时加入上述硝酸盐溶液和碱溶液,控制整个过程为4小时,ph控制7-8。形成的沉淀经过经过过滤、水洗涤至中性或滤液中检测达不到na
+
、110℃干燥8h、500℃焙烧5h、压片成形等步骤得到脱氧催化剂c-7。
86.实施例8
87.称取3.53g偏钛酸、29.41g氢氧化铝干胶,加水1l配制成氢氧化铝干胶悬浊液配制,在60℃下搅拌并使其分散均一。称取16.67g硝酸铜、81.14g乙酸锰、92.30g硝酸锌、74.19g硝酸镁,与3l水混合并搅拌均匀。称取640.02g碳酸钠、106.67g氢氧化钠,与3l水混合并搅拌均匀。在搅拌条件下,向氢氧化铝干胶悬浊液中同时加入上述硝酸盐溶液和碱溶液,控制整个过程为4小时,ph控制7-8。形成的沉淀经过经过过滤、水洗涤至中性或滤液中检测达不到na
+
、110℃干燥8h、540℃焙烧4 h、压片成形等步骤得到脱氧催化剂c-8。
88.实施例9
89.称取5.88g偏钛酸、22.06g氢氧化铝干胶,加水1l配制成氢氧化铝干胶悬浊液配制,在60℃下搅拌并使其分散均一。称取23.82g硝酸铜、64.92g乙酸锰、1110.76g硝酸锌、74.19g硝酸镁,与3l水混合并搅拌均匀。称取640.02g碳酸钠、106.67g氢氧化钠,与3l水混合并搅拌均匀。在搅拌条件下,向氢氧化铝干胶悬浊液中同时加入上述硝酸盐溶液和碱溶液,控制整个过程为4小时,ph控制7-8。形成的沉淀经过经过过滤、水洗涤至中性或滤液中检测达不到na
+
、110℃干燥10h、540℃焙烧4h、压片成形等步骤得到脱氧催化剂c-9。
90.以上实施制备催化剂的组成为cuo-mn3o
4-zno-mgo-tio
2-al2o3,具体物理组成性质,如表1所示。
91.表1实施制备催化剂物理组成性质
92.催化剂c-1c-2c-3c-4c-5c-6c-7c-8c-9cuo,m%2233557710mn3o4,m%503540253545402520zno,m%101215201220102530mgo,m%201820172010152020tio2,m%132532335al2o3,m%173020302518252015
93.将实施例制备得到的催化剂进行催化活性评价。反应体系如下:
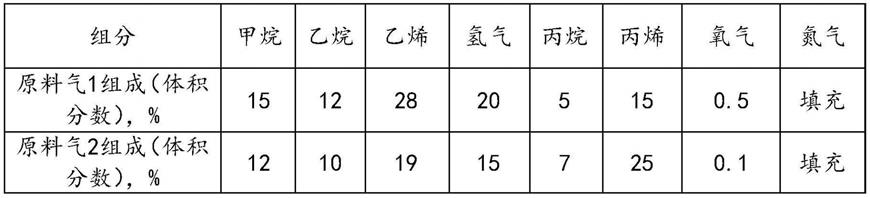
94.原料气采用配制模拟催化裂解气,如表2所示。
95.表2评价原料气组成
[0096][0097]
评价装置为列管式微型固定床,管内径为24mm,将催化剂压片整形3mm颗粒,催化剂采用密相装填50ml,高径比为4。在使用前均进行氢气干燥和还原,氢气含量99%,条件为:体积空速为600h-1
,250℃还原4h。氧含量检测采用便携式在线检测仪,以原料气1为评价原料,催化剂性能表见表3;以原料气2为评价原料,催化剂性能表见表4。
[0098]
表3原料气1为原料不同催化剂的反应条件和结果
[0099][0100][0101]
表4原料气2为原料不同催化剂的反应条件和结果
[0102][0103]
从表3和表4中可以看出,本发明所述脱氧催化剂,在高含量氢气和烯烃原料气下,反应温度较低,处理氧含量高,脱氧精度高,没有烯烃损失,此外在原料1和c-6催化剂基础进行稳定性考察,在体积空速3000h-1
,反应温度140℃及反应压力1.5mpa条件下,连续稳定运行1000h,脱后残氧含量为0.1x10-6
,且没有烯烃损失,说明本发明催化剂稳定性较高。
[0104]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应
视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1