一种氧化膦聚合物负载型催化剂及其制备方法和应用
1.本发明属于多相催化领域,具体涉及一种氧化膦聚合物负载型催化剂及其制备方法和应用。
背景技术:
2.膦配体在均相过渡金属催化的偶联反应、加氢反应、氢甲酰化反应、硅氢加成反应和co2加成等反应中具有重要的应用,通过合理的设计和修饰膦配体的电子效应和立体结构,可有效地调控目标产物的收率和选择性。膦配体与过渡金属组成的络合物催化剂普遍具有反应条件温和,活性和选择性高,副反应少等优点。但是均相催化剂分离回收困难,催化剂流失严重并且也容易造成最终产品中均相催化剂污染的问题,这些缺陷大大限制了均相催化剂在工业生产中的大规模应用。相比较而言多相催化剂与反应物料及产品的分离就容易的多,并且具有长程稳定性,因而多相催化剂也一直是工业催化剂的主流。如何制备出兼具有多相催化和均相催化双重优势的高效催化剂一直是化学工作者们努力追求的目标。
3.另一方面,传统的膦配体如三苯基膦等在使用过程中会被反应系统中的微量氧气和水分氧化,导致催化性能下降,往往需要补加三苯基膦以维持催化性能。会导致工艺操作复杂,贵金属流失等等一系列的问题。仲膦氧化物(secondary phosphine oxide,简称spo)配体是较强的给电子配体,比传统的三苯基膦配体在空气氛围下更稳定,更易于合成。本质上,spo 配体存在着五价氧化膦和三价亚磷酸的平衡。spo一般倾向于以五价磷的状态存在,但是当与金属配位后,会转化为三价亚磷酸的状态(chemcatchem, 2020,12,3982-3994)。
4.近年来,多孔有机材料的设计和合成逐渐成为微孔材料研究领域新的热点之一,多孔有机聚合物材料(pops)材料的迅猛发展为催化剂的多相化及回收使用提供了契机。
技术实现要素:
5.为了解决上述问题,本发明的目的在于提供一种氧化膦聚合物负载型催化剂及其制备方法和应用。
6.本发明的技术方案为:
7.一种氧化膦聚合物负载型催化剂,其特征在于:所述负载型催化剂以金属 rh、co、ir、ru、pt、pd和fe中的一种或两种以上作为活性组分,以氧化膦聚合物作为载体;
8.氧化膦聚合物由含有烯烃基的仲膦氧化物配体自聚而成、或者由含有烯烃基的仲膦氧化物配体与含有烯烃基第二组份共聚而成。
9.所述含有烯烃基的仲膦氧化物配体为下述中的一种或二种以上:
[0010][0011]
含有烯烃基的第二组分选自下述中的一种或二种以上:
[0012]
[0013]
[0014]
[0015][0016]
所述的氧化膦聚合物载体具有多级孔结构,比表面积为10-3000m2/g,优选范围为20-1000m2/g,孔容为0.1-10.0cm3/g,优选为0.2-2.0cm3/g,孔径分布在0.01-100.0nm,优
选为0.1-5.0nm;
[0017]
催化剂中活性组分金属担载量范围为0.01-10wt%,优选范围为0.1-3wt%。
[0018]
所述聚合用到的含有烯烃基的仲膦氧化物配体和/或含有烯烃基的第二组分烯烃基优选为乙烯基官能团的物质。
[0019]
1)氧化膦聚合物的制备过程为:
[0020]
将含有烯烃基的仲膦氧化物配体溶解后、或将含有烯烃基的仲膦氧化物配体和含有烯烃基第二组份溶解混合后,经自由基引发剂引发有机膦配体中的烯烃基发生聚合反应,生成具有多级孔结构氧化膦聚合物载体;
[0021]
氧化膦聚合物负载型催化剂制备方法为:活性金属组分的前驱体与氧化膦聚合物载体在溶剂中充分搅拌配位,活性金属组分与氧化膦聚合物载体中裸露的p形成牢固的化学键,蒸去溶剂后,得到氧化膦聚合物负载型催化剂。
[0022]
氧化膦聚合物负载型催化剂具体制备步骤为:
[0023]
a)惰性气体气氛273-473k(优选为298-453k)下,在溶剂中加入含有烯烃基的仲膦氧化物配体、或加入含有烯烃基的仲膦氧化物配体和含有烯烃基第二组份,再加入自由基引发剂,将混合物搅拌0.1-100小时得预聚体溶液,优选的搅拌时间范围为0.1-20小时;
[0024]
b)将步骤a)制得的预聚体混合溶液转移至聚合反应器中,采用本体聚合、溶液聚合、悬浮聚合或乳液聚合等方法中的一种或二种以上,聚合时间1-100 小时进行聚合反应,得到一种氧化膦聚合物;
[0025]
c)将步骤b)得到的氧化膦聚合物,273-403k(优选为303-453k)下除溶剂,即得到具有多级孔结构的氧化膦聚合物,即氧化膦聚合物负载型催化剂的载体;
[0026]
d)惰性气体气氛273-473k(优选为298-453k)下,在含有活性金属组分前驱体的溶剂中,加入步骤c)得到的氧化膦聚合物载体,搅拌0.1-100小时,优选搅拌时间范围0.1-20小时,之后,273-403k(优选为303-453k)下除溶剂,得到氧化膦聚合物负载型催化剂;活性金属在前驱体溶液中的浓度范围为0.001-1mol l-1。
[0027]
步骤a)和d)中所述的溶剂为甲醇、乙醇、二氯甲烷、三氯甲烷、苯、甲苯、二甲苯、水或四氢呋喃中一种或两种以上;
[0028]
步骤a)中所述的自由基引发剂为叔丁基过氧化氢、偶氮二异丁腈、偶氮二异庚腈、过氧化环己酮或过氧化二苯甲酰中的一种或两种以上。
[0029]
步骤a)中所述的含有烯烃基的仲膦氧化物配体和含有烯烃基第二组份的摩尔比为0.01:1-100:1,优选为1:1-1:10,含有烯烃基的仲膦氧化物配体与自由基引发剂的摩尔比为500:1-10:1,优选为100:1-10:1;聚合成有机聚合物前,含有烯烃基的仲膦氧化物在溶剂中的浓度范围为0.01-1000g/l,优选为 0.1-10g/l;步骤a)、b)和d)中所述惰性气体分别选自ar、he、n2和co2 中的一种或两种以上。
[0030]
所述的活性组分为rh、co、ir、ru、pt、pd或fe中的一种或两种以上,其中rh的前驱体为rhh(co)(pph3)3、rh(co)2(acac)、rhcl3、rh(ch3coo)2中的一种或两种以上;co的前驱体为co(ch3coo)2、co(co)2(acac)、co (acac)2、cocl2中的一种或两种以上;ir的前驱体为ir(co)3(acac)、 ir(ch3coo)3、ir(acac)3、ircl4中的一种或两种以上;ru的前驱体为二氯(环辛基-1,5-二烯)钌(ii)、rucl3、ru(acac)3、十二羰基三钌、[ruar2(benzene)]2、 [ruar2(p-cymene)]2,[ruar2(mesitylene)]2、[(π-ally)ru(cod)]2、 [(π-ally)ru(nbd)]2中的一种或
两种以上;pt的前驱体为pt(acac)2、ptcl4、 ptcl2(nh3)2中的一种或两种以上;pd的前驱体为pd(ch3coo)2、pd(acac)2、 pdcl2、pd(pph3)4、pdcl2(ch3cn)2中的一种或两种以上;fe的前驱体为 fe(acac)3、fecl3、fecl2、fes、二茂铁、九羰基二铁中的一种或两种以上,催化剂中金属担载量范围为0.01-10wt%,优选为0.1-3wt%。所述氧化膦聚合物负载型催化剂在烯烃氢甲酰化反应、氢甲胺化、氢羧基化或醇类羰基化反应中的应用;其中原料烯烃碳数范围为c2-c30,原料醇类的碳数范围为 c1-c10。
[0031]
本发明的反应原理:
[0032]
本发明创造性地将烯烃基引入到仲膦氧化物有机单体上,利用单体分子上的烯烃基的聚合反应制备出氧化膦体相浓度高,孔隙发达的功能聚合物载体。氧化膦载体聚合物具有载体和配体的双重作用,氧化膦聚合物体相中的p与金属配位后可形成适用于烯烃氢甲酰化反应、氢甲胺化、氢羧基化、醇类羰基化反应的高性能催化剂。本专利所保护的催化剂本是非均相的,催化剂易于从反应体系中分离出来,并且催化剂是乙烯基聚合方案制备的,催化剂具有长程稳定性,适合于工业应用。
[0033]
本发明的有益效果为:
[0034]
传统的膦配体如三苯基膦等在使用过程中会被反应系统中的微量氧气和水分氧化,导致催化性能下降,往往需要补加三苯基膦以维持催化性能。会导致工艺操作复杂,贵金属流失等等一系列的问题。本发明提供的仲膦氧化物负载型催化剂从根本上解决这个问题,仲膦氧化物对水分和氧气稳定,操作条件没有那么苛刻,并且得益于仲膦氧化物与金属配位后,p上所连接的羟基与金属和p原子的协同作用,与传统的三苯基膦作为配体的催化剂相比,本专利提供的催化剂活性更高。
[0035]
本发明负载型催化剂适用于固定床,浆态床,釜式反应器和滴流床等反应器中。本发明提供的氧化膦负载型催化剂可应用于烯烃氢甲酰化反应、氢甲胺化、氢羧基化、醇类羰基化反应等反应中,与传统的三苯基膦作为配体的催化剂相比,催化活性更高;催化剂对空气和水分稳定,操作条件不需要太苛刻。并且催化剂是非均相的,基于本专利涉及的氧化膦负载型催化剂进行氢甲酰化的方法绿色环保,污染物排放少。
附图说明
[0036]
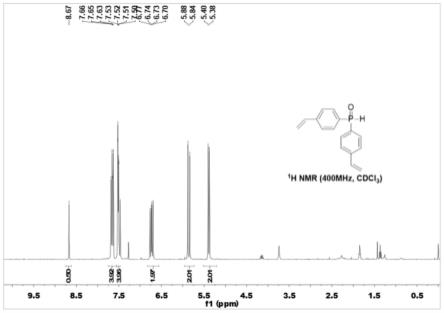
图1是二苯乙烯基氧化膦(实施例1中的a单体)的1h nmr谱图。
[0037]
图2是二苯乙烯基氧化膦(实施例1中的a单体)的
13
c nmr谱图。
[0038]
图3是二苯乙烯基氧化膦(实施例1中的a单体)的
31
p nmr谱图。
[0039]
图4为cpol-1spo10dvb物理吸附图。
[0040]
图5为cpol-1spo10dvb孔径分布图。
[0041]
图6为rh/cpol-1spo10dvb催化剂的热重曲线。
具体实施方式
[0042]
下述实施例对本发明进行更好的说明,但不限制本发明所要保护的范围。
[0043]
实施例1
[0044]
a单体具体制备步骤:273k氩气保护下,9g对氯苯乙烯溶于50ml 2
‑ꢀ
甲基四氢呋喃,搅拌均匀待用。1.7g镁屑放入烧瓶中,将烧瓶温度升至333k,滴加5ml对氯苯乙烯和2-甲
基四氢呋喃的混合溶液,格式试剂引发后(反应溶液颜色变成墨绿色,剧烈沸腾)继续滴加剩余的混合溶液,维持滴加温度为65℃。滴加结束后保温1小时得对氯苯乙烯的格式试剂溶液。后降温至0摄氏度,加入4.5g亚磷酸二乙酯与50毫升2-甲基四氢呋喃的混合溶液,滴加完成后继续反应1小时。加入10ml饱和nh4cl溶液湮灭反应,混合液分为两层,将上层油层取出,60℃下蒸馏除去溶剂得淡黄色油状液体,加入10ml正庚烷将混合溶剂加热至60℃充分溶解,后降温至0℃重结晶并干燥后即可得到二苯乙烯基氧化膦(权利要求中的配体a)6.05g(收率73%左右,产品经核磁及高分辨质谱确认),附图1-3分别为二苯乙烯基氧化膦a的核磁1h、
13
c和
31
p谱图。
[0045]
二苯乙烯基氧化膦多孔有机聚合物的制备:在298k和惰性气体氩气保护氛围下,将5.0克二苯乙烯基氧化膦配体a和50.0g第二组分二乙烯基苯 (权利要求中的配体l1,cas号1321-74-0)溶于550.0ml四氢呋喃溶剂中,向上述溶液中加入0.01克自由基引发剂偶氮二异丁腈,搅拌2小时得预聚体。将预聚体转移至高压釜中,于373k和惰性气体氩气保护氛围下利用本体聚合法聚合24h。待聚合釜冷却至室温,室温条件真空抽走溶剂,即得到由二苯乙烯基氧化膦与二乙烯基苯共聚的聚合物cpol-1spo10dvb,产率100%。附图4为cpol-1spo10dvb的n2物理吸附曲线,附图5为cpol-1spo10dvb的孔径分布图,从图中可以看出cpol-1spo10dvb呈现多级孔结构的吸附曲线,比表面积905m2/g,孔径主要分布在0.4-5nm。
[0046]
氧化膦聚合物负载型高分散rh金属催化剂:称取9.0毫克(1,5-环辛二烯)2,4-戊二酮铑(i)(cas号12245-39-5)溶于10.0ml四氢呋喃溶剂中,加入1.0克上述制得的cpol-1spo10dvb聚合物,298k搅拌5小时,在298k 氩气氛围下继续搅拌5小时,318k条件下真空抽除溶剂,即获得氧化膦聚合物自负载型的高分散rh基催化剂rh/cpol-1spo10dvb,实测rh负载量0.29%。stem电镜观察金属rh处于单分散的状态。图6为 rh/cpol-1spo10dvb催化剂的热重曲线,从图中可以看出催化剂在430℃以上才出现分解的失重峰。
[0047]
实施例2
[0048]
在实施例2中,除了不添加50.0g第二组分二乙烯基苯外,其余的合成过程和条件与实施例1相同。
[0049]
实施例3
[0050]
在实施例3中,除了用50.0g第二组分三(4-乙烯基苯基)膦代替二乙烯基苯外,其余的合成过程和条件与实施例1相同。
[0051]
实施例4
[0052]
在实施例4中,除了用将聚合时间由24小时调整为48小时外,其余的方法和催化剂合成过程和条件与实施例1相同。
[0053]
实施例5
[0054]
在实施例5中,除了用相同摩尔数co(acac)2替代(1,5-环辛二烯)2,4
‑ꢀ
戊二酮铑(i)外,其余合成过程和条件与实施例1相同,可得高分散型co基催化剂。
[0055]
实施例6
[0056]
在实施例6中,除了用相同摩尔数ircl3替代(1,5-环辛二烯)2,4-戊二酮铑(i)外,其余合成过程和条件与实施例1相同,可得高分散型ir基催化剂。
[0057]
实施例7
[0058]
在实施例7中,除了用相同摩尔数十二羰基三钌替代(1,5-环辛二烯)2,4
‑ꢀ
戊二酮
铑(i)外,其余合成过程和条件与实施例1相同。
[0059]
实施例8
[0060]
在实施例8中,除了用相同摩尔数二茂铁替代(1,5-环辛二烯)2,4-戊二酮铑(i)外,其余合成过程和条件与实施例1相同。
[0061]
实施例9
[0062]
在实施例9中,除了用相同摩尔数三(4-乙烯基苯基)膦代替二苯乙烯基氧化膦单体a外,其余的合成过程和条件与实施例1相同,可得 rh/cpol-10dvb1pph3的对比催化剂。
[0063]
实施例10
[0064]
在实施例10中,除了用相同摩尔数配体c代替二苯乙烯基氧化膦单体a外,其余的合成过程和条件与实施例1相同。
[0065]
实施例11
[0066]
在实施例11中,除了用相同摩尔数配体f代替二苯乙烯基氧化膦单体 a外,其余的合成过程和条件与实施例1相同。
[0067]
实施例12
[0068]
在实施例12中,除了用相同摩尔数配体j代替二苯乙烯基氧化膦单体 a外,其余的合成过程和条件与实施例1相同。
[0069]
实施例13
[0070]
在实施例13中,除了用相同摩尔数配体n代替二苯乙烯基氧化膦单体 a外,其余的合成过程和条件与实施例1相同。
[0071]
实施例14
[0072]
在实施例14中,除了用相同摩尔数配体r代替二苯乙烯基氧化膦单体 a外,其余的合成过程和条件与实施例1相同。
[0073]
实施例15
[0074]
在实施例15中,除了用相同摩尔数配体t代替二苯乙烯基氧化膦单体 a外,其余的合成过程和条件与实施例1相同。
[0075]
实施例16
[0076]
在实施例16中,除了用相同摩尔数配体v代替二苯乙烯基氧化膦单体 a外,其余的合成过程和条件与实施例1相同。
[0077]
实施例17
[0078]
将实施例1至实施例16制备的催化剂2g分别装入到固定床反应器中,催化剂床层的两端装入石英砂。通入反应混合气(h2:co:c2h4=1:1:1, v/v/v),在393k,1.0mpa,反应混合气空速4000h-1
条件下进行氢甲酰化反应。反应产物经一个装有60ml冷却的去离子水的收集罐吸收收集,反应产物丙醛全部溶于收集罐的水中。所获得水溶液采用配有hp-5毛细管柱和 fid检测器的hp-7890n气相色谱分析,采用乙醇作内标。经水吸收后反应尾气采用配有porapak-qs柱和tcd检测器的hp-7890n气相色谱进行在线分析。反应结果列于表1。
[0079]
表1.实施例1-实施例16催化剂比表面积、孔径分布和乙烯氢甲酰化性能
[0080][0081][0082]
实施例9制备的是三苯基膦配位的rh催化剂(对比例),实施例1制备的是氧化膦配位的rh催化剂,从乙烯氢甲酰化的反应数据可以看出,氧化膦配位的rh催化剂乙烯氢甲酰化活性更高,醛选择性也更好。实施例5、6、7、8活性金属组分分别为co、 ir、ru、fe,反应性能低于活性金属为rh的催化剂。当活性中心为rh时,从表中数据可以看出,氧化膦配位的rh催化剂性能要比三苯基膦配位的rh催化剂活性更高,同时醛的选择性也好一些。
[0083]
实施例18
[0084]
用相同摩尔数醋酸钯替代(1,5-环辛二烯)2,4-戊二酮铑(i)外,其余催化剂合成过程和条件与实施例1相同,可得氧化膦负载的pd催化剂,实测pd 负载量为0.3%。
[0085]
100ml高压釜中,加入15mg氧化膦负载的pd催化剂,nh4cl 1mmol,反应物己烯5mmol,溶剂n甲基吡咯烷酮(nmp)5ml,后充入3mpa co进行氢甲胺化反应。
[0086]
反应温度24小时,反应温度413k。己烯转化率88.6%,产品选择性 99.8%。
[0087]
实施例19
[0088]
100ml高压釜中,加入实施例1中的催化剂0.5g,对甲基苯磺酸 1.0mmol,反应物环己烯6mmol,碘甲烷3mmol,纯净水50mmol,后充入 2mpa co进行氢羧基化反应。
[0089]
反应时间24小时,反应温度455k,环己烯转化率96.7,产品选择性 90.5%。
[0090]
实施例20
[0091]
将实施例1和实施例9制备的催化剂5g放入石英管中,80℃,空气压力1.1bar,空速1000h-1处理5小时,得处理后的催化剂。将处理后的催化剂2g分别装入到固定床反应器中,催化剂床层的两端装入石英砂。通入反应混合气(h2:co:c2h4=1:1:1,v/v/v),在393k,1.0mpa,反应混合气空速4000h-1
条件下进行氢甲酰化反应。反应产物经一个装有60ml冷却的去离子水的收集罐吸收收集,反应产物丙醛全部溶于收集罐的水中。所获得水溶液采用配有hp-5毛细管柱和fid检测器的hp-7890n气相色谱分析,采用乙醇作内标。经水吸收后反应尾气采用配有porapak-qs柱和tcd检测器的hp-7890n气相色谱进行在线分析。反应结果列于表2。
[0092]
表2.实施例1和实施例9空气处理后催化剂比表面积和乙烯氢甲酰化性能
[0093][0094]
实施例9制备的是三苯基膦配位的rh催化剂(对比例),实施例1制备的是氧化膦配位的rh催化剂,空气处理后,实施例9乙烯氢甲酰化性能发生了明显的下降(结合表1数据进行对比),可能是由于三苯基膦空气中不稳定造成的,而实施例1催化剂处理后乙烯氢甲酰化性能未发生明显的变化,这说明氧化膦催化剂对空气和水分稳定,催化剂稳定性更好,操作条件不需要太苛刻,更有利于工业生产。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1