一种自组装铁单原子类芬顿异相催化剂、制备方法及应用
1.本发明涉及水处理技术领域,具体为一种自组装铁单原子类芬顿异相催化剂、制备方法及应用。
背景技术:
2.随着时代需求高速发展的制药、印染、纺织、农业等行业,造福人类的同时大量难降解有机污染物被排放进入自然水体,对水体生态系统和人类健康产生极大的危害。目前亟需寻求高效、绿色、经济的水处理技术去应对水体污染这一挑战,其中,基于单过氧硫酸盐(pms)的高级氧化水处理技术受到广泛关注。单过氧硫酸盐具有化学性质稳定、储运安全、成本低的优点,经过活化产生具有强氧化性的自由基攻击有机污染物,或通过非自由基途径具更高选择性降解目标污染物,达到水体净化的效果。然而,活化单过氧硫酸盐的均相催化剂普遍存在成本高、污染大、性能差、难以循环利用等缺点,因此,非均相金属催化剂常应用于pms的活化,但其亦较多存在金属利用率低、金属离子浸出浓度大、催化活性中心少、催化性能较低等问题。铁是自然界中广泛存在的金属元素,排入自然水体中对生物和生态环境相对是绿色有好的。铁金属单原子催化剂是异相催化剂中的一类,具有金属原子利用率大,催化活性中心分离且明确的优势。对于金、铂等贵金属催化剂而言,金属中心分离使原子利用率接近100%大大提高了其经济性。
技术实现要素:
3.本发明的目的在于提供一种自组装铁单原子类芬顿异相催化剂、制备方法及应用,其利用铁金属单原子异相催化剂的优点,采用低成本的合成材料,提高目标催化剂的催化性能,同时具有催化剂的稳定性较高、金属离子浸出浓度低,反应后易于回收,以解决上述背景技术中提出的现有催化剂存在不足之处。
4.为实现上述目的,本发明提供如下技术方案:
5.一种自组装铁单原子类芬顿异相催化剂的制备方法,包括以下步骤:
6.1)将无水乙醇加入去离子水中,混合均匀后,得到溶液a;
7.2)将无水柠檬酸溶解于溶液a中,磁力搅拌器上搅拌均匀后,得到悬浊液b;
8.3)往步骤2中制备而得的悬浊液b中加入三价铁源,磁力搅拌器上剧烈搅拌,得到悬浊液c;
9.4)往步骤3中所得的悬浊液c中加入三聚氰胺,超声均匀后,得到黏稠状浆体d;
10.5)将步骤4中所得浆体d置于水浴锅中,加热搅拌,直至蒸干,研磨至粉状固体e;
11.6)将步骤5中所得的粉状固体e放入管式电阻炉中,在氮气氛围中程序升温煅烧,自然冷却至室温,研磨,即得到自组装铁单原子类芬顿异相催化剂。
12.优选的,所述步骤1中,无水乙醇为分析纯级,去离子水为实验室超纯水仪器制备,无水乙醇和去离子水的体积为:无水乙醇:去离子水=30-60ml:30-60ml;步骤1中,搅拌工具为玻璃棒,混合均匀的温度为25-35℃。
13.优选的,所述步骤2中,无水柠檬酸是分析纯级,投加摩尔数为5-15mmol;步骤2中,搅拌的温度为25-35℃,搅拌的速率为350-500r/min,搅拌的时间为5-15min。
14.优选的,所述步骤3中,三价铁源为六水合三氯化铁,三价铁离子投加摩尔数为0.1-0.6mmol;步骤3中,搅拌的温度为25-35℃,搅拌的速率为500-700r/min,搅拌的时间为5-15min。
15.优选的,所述步骤4中,三聚氰胺为分析纯级,投加摩尔数为30-200mmol;步骤4中,超声的频率为40khz,超声的温度为30-60℃,超声的时间为15-45min。
16.优选的,所述步骤5中,水浴锅中搅拌的温度为70-90℃,搅拌的速率为500-700r/min,搅拌的时间为20-120min。
17.优选的,所述步骤6中,营造氮气氛围的氮气纯度为高纯氮(≥99.9%),气体流速为50-200ml/min;步骤6中,管式炉的第一阶段升温速率为2.3-5℃/min,升温至500-600℃,煅烧时间为0.5-1.5h;步骤6中,管式炉的第二阶段升温速率为2.3-5℃/min,升温至700-900℃,煅烧时间为2-4h。
18.一种自组装铁单原子类芬顿异相催化剂,其是所述的制备方法经过自组装过程得到的自组装铁单原子类芬顿异相催化剂。
19.一种自组装铁单原子类芬顿异相催化剂的应用,将所述自组装铁单原子类芬顿异相催化剂,应用于处理水体中难降解有机污染物。
20.优选的,所述难降解有机污染物为双酚a(bpa)、磺胺甲恶唑(smx)、4-氯苯酚(4-cp)、2-氯苯酚(2-cp)中的至少一种。
21.与现有技术相比,本发明的有益效果是:
22.1、本发明提供的自组装铁单原子类芬顿异相催化剂及其制备方法,利用铁金属单原子异相催化剂的优点,采用低成本的合成材料,提高目标催化剂的催化性能,同时具有催化剂的稳定性较高、金属离子浸出浓度低,反应后易于回收且制备方法简便;
23.2、本发明催化剂能够高效降解水体中难降解有机污染物,大大缩短了经典芬顿反应降解有机物的反应时间,提高处理效率;
24.3、本发明催化剂在较高浓度无机阴离子和天然有机物水体中,对难降解有机污染物仍有很好的去除效果;
25.4、本发明催化剂在整个催化过程中铁金属离子溶出浓度低,具有环境友好的优点;
26.5、本发明催化剂进行的固定床流动实验能够维持长时间较高的有机污染物去除率,催化剂具有很好的稳定性;
27.6、本发明催化剂为固体催化剂,反应后便于与水体分离,避免造成“二次污染”;
28.7、本发明催化剂合成条件温和,操作步骤简单,制备过程耗时短,所需合成原料廉价经济。
附图说明
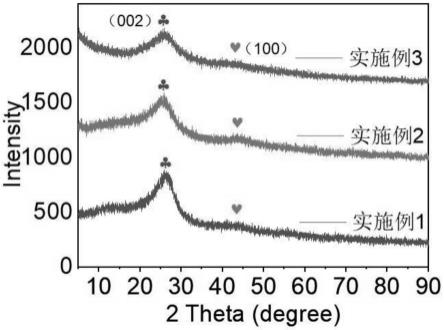
29.图1为实施例1、2、3中制备所得的fe-sac-nc的x射线粉末衍射图;
30.图2为实施例3中制备所得的fe-sac-nc和实施例1、2中制备所得的fe-sac-nc对双酚a的降解曲线图;
31.图3为实施例3中制备所得的fe-sac-nc在较高浓度水体阴离子体系中对bpa的降解曲线图;
32.图4为实施例3中制备所得的fe-sac-nc在较高浓度腐殖酸体系中对bpa的降解曲线图;
33.图5为实施例3中制备所得的fe-sac-nc对bpa、smx、4-cp、2-cp的降解曲线图;
34.图6为实施例3中制备所得的fe-sac-nc和实施例1中制备所得的fe-sac-nc在固定流动反应器中对bpa的稳定性降解评价图。
具体实施方式
35.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
36.请参阅图1-6,本发明提供如下几种具体的技术方案。
37.一种自组装铁单原子类芬顿异相催化剂的制备方法,包括以下步骤:
38.1)将无水乙醇加入去离子水中,混合均匀后,得到溶液a;
39.2)将无水柠檬酸溶解于溶液a中,磁力搅拌器上搅拌均匀后,得到悬浊液b;
40.3)往步骤2中制备而得的悬浊液b中加入三价铁源,磁力搅拌器上剧烈搅拌,得到悬浊液c;
41.4)往步骤3中所得的悬浊液c中加入三聚氰胺,超声均匀后,得到黏稠状浆体d;
42.5)将步骤4中所得浆体d置于水浴锅中,加热搅拌,直至蒸干,研磨至粉状固体e;
43.6)将步骤5中所得的粉状固体e放入管式电阻炉中,在氮气氛围中程序升温煅烧,自然冷却至室温,研磨,即得到自组装铁单原子类芬顿异相催化剂。
44.实施例一:
45.本实施例提供的自组装铁单原子类芬顿异相催化剂的制备方法,包括如下步骤:
46.1)将50ml无水乙醇加入50ml去离子水中,在温度30℃下,通过玻璃棒搅拌混合均匀后,得到溶液a;
47.2)将10mmol无水柠檬酸加入溶液a中,在温度30℃下,置于磁力搅拌器上,转速为450r/min搅拌10min,得到悬浊液b;
48.3)往步骤2中制备而得的悬浊液b中加入0.1mmol六水合三氯化铁配制的三价铁离子(fe3+),在温度30℃下,置于磁力搅拌器上剧烈搅拌,转速为700r/min,搅拌的时间为10min,得到悬浊液c;
49.4)往步骤3中所得的悬浊液c中加入30mmol三聚氰胺,超声的频率是40khz,超声的温度为40℃、时间是30min,待超声均匀后得到黏稠状浆体d;
50.5)将步骤4中所得浆体d置于水浴锅中,以80℃水浴一边加热一边磁力搅拌,磁力搅拌转速为700r/min,加热蒸干40min,研磨至粉状固体e;
51.6)将步骤5中所得的粉状固体e放入管式电阻炉中,在氮气氛围中经过两个阶段程序升温煅烧,第一个煅烧阶段为:升温速率为5℃/min,升温至550℃,煅烧时间为1h;紧接着进入第二个煅烧程序:升温速率为5℃/min,升温至800℃,煅烧时间为2h。程序结束后,自然
冷却至室温,研磨,即得到自组装铁单原子类芬顿异相催化剂fe-nc-sac。
52.实施例二:
53.本实施例提供的自组装铁单原子类芬顿异相催化剂的制备方法,包括如下步骤:
54.1)将50ml无水乙醇加入50ml去离子水中,在温度30℃下,通过玻璃棒搅拌混合均匀后,得到溶液a;
55.2)将10mmol无水柠檬酸加入溶液a中,在温度30℃下,置于磁力搅拌器上,转速为450r/min搅拌10min,得到悬浊液b;
56.3)往步骤2中制备而得的悬浊液b中加入0.6mmol六水合三氯化铁配制的三价铁离子(fe3+),在温度30℃下,置于磁力搅拌器上剧烈搅拌,转速为700r/min,搅拌的时间为10min,得到悬浊液c;
57.4)往步骤3中所得的悬浊液c中加入200mmol三聚氰胺,超声的频率是40khz,超声的温度为40℃、时间是30min,待超声均匀后得到黏稠状浆体d;
58.5)将步骤4中所得浆体d置于水浴锅中,以80℃水浴一边加热一边磁力搅拌,磁力搅拌转速为700r/min,加热蒸干40min,研磨至粉状固体e;
59.6)将步骤5中所得的粉状固体e放入管式电阻炉中,在氮气氛围中经过两个阶段程序升温煅烧,第一个煅烧阶段为:升温速率为5℃/min,升温至550℃,煅烧时间为1h;紧接着进入第二个煅烧程序:升温速率为5℃/min,升温至800℃,煅烧时间为2h。程序结束后,自然冷却至室温,研磨,即得到自组装铁单原子类芬顿异相催化剂fe-nc-sac。
60.实施例三:
61.本实施例提供的自组装铁单原子类芬顿异相催化剂的制备方法,包括如下步骤:
62.1)将50ml无水乙醇加入50ml去离子水中,在温度30℃下,通过玻璃棒搅拌混合均匀后,得到溶液a;
63.2)将10mmol无水柠檬酸加入溶液a中,在温度30℃下,置于磁力搅拌器上,转速为450r/min搅拌10min,得到悬浊液b;
64.3)往步骤2中制备而得的悬浊液b中加入0.3mmol六水合三氯化铁配制的三价铁离子(fe3+),在温度30℃下,置于磁力搅拌器上剧烈搅拌,转速为700r/min,搅拌的时间为10min,得到悬浊液c;
65.4)往步骤3中所得的悬浊液c中加入90mmol三聚氰胺,超声的频率是40khz,超声的温度为40℃、时间是30min,待超声均匀后得到黏稠状浆体d;
66.5)将步骤4中所得浆体d置于水浴锅中,以80℃水浴一边加热一边磁力搅拌,磁力搅拌转速为700r/min,加热蒸干40min,研磨至粉状固体e;
67.6)将步骤5中所得的粉状固体e放入管式电阻炉中,在氮气氛围中经过两个阶段程序升温煅烧,第一个煅烧阶段为:升温速率为5℃/min,升温至550℃,煅烧时间为1h;紧接着进入第二个煅烧程序:升温速率为5℃/min,升温至800℃,煅烧时间为2h。程序结束后,自然冷却至室温,研磨,即得到自组装铁单原子类芬顿异相催化剂fe-nc-sac。
68.试验例1:测试实施例1、2、3材料的表征
69.图1所示为实施例1、2、3制备所得的自组装铁单原子类芬顿异相催化剂fe-nc-sac的x射线粉末衍射。三个实施例均未形成fe的金属衍射峰,这表明三个实施例合成的铁单原子催化剂引入的fe元素是孤立均匀分散的,没有形成铁金属纳米颗粒或者金属氧化物,fe
在催化剂中与催化剂的基底元素配位孤立存在。实施例1、2、3制备的fe-nc-sac是碳基催化剂,一般而言,碳材料在2θ为25
°
和43
°
附近会有两个衍射峰,分别对应碳材料的(002)和(100)晶面。在三个实施例制备的fe-nc-sac的xrd图谱中,在2θ为24
°
和44
°
附近显示两个宽化的衍射峰,这意味着该催化剂形成了具有较低石墨化程度的结晶碳,因为催化剂的合成前驱体中加入三聚氰胺,引入了氮元素,所以在碳晶格中掺入的氮元素使晶体发生略微的畸变,产生的无序结构。实施例3的(002)晶型相对实施例1、2的峰强更弱,且实施例3的(100)晶型几乎消失,峰宽化程度最大,因此,实施例3的结晶碳中引入的氮元素更多。由于氮元素的掺入,与fe更好地配位,使fe元素更稳定固定在催化剂中,不仅提高催化剂稳定性,且增加fe金属的负载量,增加催化活性中心。
70.试验例2应用实验
71.试验过程1:称取5mg上述实施例3制备所得的fe-nc-sac催化剂、实施例1制备所得的fe-nc-sac催化剂、实施例2制备所得的fe-nc-sac催化剂分别投加到50ml含难降解有机污染物的溶液中,经过15min超声和15min磁力搅拌均匀反应体系,反应在室温中进行,加入0.5mm单过氧硫酸盐(pms)启动催化反应,在不同的反应时间点取样测定体系中难降解有机污染物的浓度,其中实施例1和实施例2制备的fe-nc-sac在活化pms类芬顿反应中的效果与实施例3相似,在此不一一赘述。
72.试验过程2:称取10mg上述实施例3制备所得的fe-nc-sac催化剂、实施例1制备所得的fe-nc-sac催化剂分别与5g洗净石英砂均匀混合,装入固定床流动反应器的石英柱中,再称取23g石英砂堵塞于石英柱两端,连接两根液体输送管道,一端为双酚a污染物,另一端为pms溶液,启动反应后,反应在室温中进行,在不同的反应时间点取样测定体系中难降解有机污染物的浓度。
73.图2为实施例3中制备所得的fe-sac-nc和实施例1、2中制备所得的fe-sac-nc对双酚a(bpa)的降解曲线图。图2为各个实施例中制备的铁单原子异相催化剂在单过氧硫酸盐类芬顿体系中降解bpa的情况。在实施例1催化剂体系中,fe-sac-nc催化单过氧硫酸盐降解bpa在150s时,去除率达到82%;实施例2、3制备的fe-sac-nc体系中,反应进行到150s的时候,对bpa的去除率效果都达到100%,并且相对经典芬顿体系降解有机污染物的反应时间短。对比实施例2和实施例3制备的fe-sac-nc对bpa的降解过程,实施例3的fe-sac-nc催化的反应速率更快,在120s的反应时间近乎完全去除bpa。以上催化效果说明,实施例3催化剂中掺入的三聚氰胺量为适宜的,氮元素配位固定的fe金属元素形成的催化活性中心的催化效果是最佳的。
74.图3为实施例3中制备所得的fe-sac-nc在较高浓度水体阴离子体系中对bpa的降解曲线图。如图3所示,1mm不同水体阴离子环境对实施例3制备的fe-sac-nc对bpa的催化降解效果影响程度不大。在1mm的氯离子(cl-)、磷酸二氢根离子(h2po4-)、硝酸根离子(no3-)体系中,fe-sac-nc对bpa的催化降解效果与fe-sac-nc的control体系(不加阴离子)对比几乎不受影响;而在碳酸根离子(co32-)体系中,fe-sac-nc对bpa的去除率降低为79%。由此可见,在大多数高浓度的水体阴离子环境中,实施例3制备的fe-sac-nc仍能够高效去除bpa。但是,control体系中fe-sac-nc对bpa的吸附率约为30%,而co32-体系中fe-sac-nc对bpa的吸附率约为10%,由于co32-影响fe-sac-nc对bpa的吸附效果从而影响其催化降解效果。以上催化效果说明,co32-吸附在催化剂上,占据了fe-sac-nc的活性位点,减少bpa与活
性位点的接触,使得催化效果降低。
75.图4为实施例3中制备所得的fe-sac-nc在较高浓度腐殖酸体系中对bpa的降解曲线图。图4展示在高浓度天然有机物腐殖酸的影响下,实施例3制备的fe-sac-nc对bpa仍具有很好的降解效果。在高浓度的腐殖酸环境中,fe-sac-nc选择性地催化降解bpa,这表明fe-sac-nc应用于实际水体中能够很好地选择性去除bpa等目标有机污染物。
76.图5为实施例3中制备所得的fe-sac-nc对bpa、smx、4-cp、2-cp的降解曲线图。反应进行到约60s时,fe-sac-nc对4-cp、2-cp和bpa的去除率近乎100%;反应进行到150s时,对smx的去除率接近90%。由上述对多种有机物的去除情况来看,fe-sac-nc表现出良好的降解效果,这说明实施例3制备的fe-sac-nc催化剂能够针对性地处理多种有机物污染的水体。
77.图6为实施例3中制备所得的fe-sac-nc和实施例1中制备所得的fe-sac-nc在固定流动反应器中对bpa的稳定性降解评价图。由固定床流动反应器的实验结果来看,实施例1制备的fe-sac-nc在反应1h对bpa的去除率约为94%,反应6h时对bpa的去除率仅为10%。实施例3制备的fe-sac-nc催化剂,从反应启动至反应15h,其对bpa的去除率都为100%,从处理的第18h开始,去除率为97%未能对bpa完全去除,反应持续至26h仍对bpa有77.6%的去除率,反应直到33h时,固定床流动反应器中的fe-sac-nc对bpa的降解效果降低到约53%。以上结果充分体现了实施例3制备所得的fe-sac-nc催化剂的稳定性优势,可以应用于实际微污染水体的处理。
78.本发明提供的催化剂、制备方法及其应用,重点是利用铁金属单原子异相催化剂的优点,采用低成本的合成材料,提高目标催化剂的催化性能,同时具有催化剂的稳定性较高、金属离子浸出浓度低,反应后易于回收且制备方法简便。
79.需要特别说明的是,在本发明记载的组分、配比及工艺参数范围内,具体选择其他的组分、配比或者取值,均可以实现本发明记载的技术效果,故不再一一将其列出。
80.尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1